




























































Lecture_4_Blast_Programming_Slide Title: Applications of Bioinformatics.pptx
- 1. College of Basic and Applied Sciences (CBAS) School of Physical and Mathematical Sciences (SPMS) 2021/2022/2nd Semester CSCD 606 Bioinformatics Lecture 4 – BLAST Programming Course Lecturer: Dr Kofi Sarpong Adu-Manu Contact Information: ksadu-manu@ug.edu.gh
- 2. O'Reilly Bioinformatics Technology - BLAST Programming 2 The BLAST algorithm
- 3. O'Reilly Bioinformatics Technology - BLAST Programming 3 What is BLAST? • Basic Local Alignment Search Tool • Calculates similarity for biological sequences. • Produces local alignments: only a portion of each sequence must be aligned. • Uses statistical theory to determine if a match might have occurred by chance.
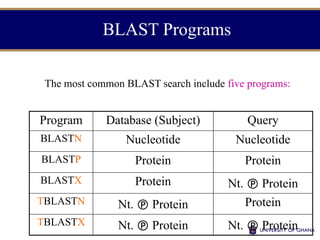
- 4. BLAST Programs The most common BLAST search include five programs: Program Database (Subject) Query BLASTN Nucleotide Nucleotide BLASTP Protein Protein BLASTX Protein Nt. Protein TBLASTN Nt. Protein Protein TBLASTX Nt. Protein Nt. Protein
- 5. BLASTN • BLASTN – The query is a nucleotide sequence – The database is a nucleotide database – No conversion is done on the query or database • DNA :: DNA homology – Mapping oligos to a genome – Annotating genomic DNA with transcriptome data from ESTs and RNA-Seq – Annotating untranslated regions
- 6. BLASTP • BLASTP – The query is an amino acid sequence – The database is an amino acid database – No conversion is done on the query or database • Protein :: Protein homology – Protein function exploration – Novel gene make parameters more sensitive
- 7. BLASTX • BLASTX – The query is a nucleotide sequence – The database is an amino acid database – All six reading frames are translated on the query and used to search the database • Coding nucleotide seq :: Protein homology – Gene finding in genomic DNA – Annotating ESTs and transcripts assembled from RNA- Seq data
- 8. TBLASTN • TBLASTN – The query is an amino sequence – The database is a nucleotide database – All six frames are translated in the database and searched with the protein sequence • Protein :: Coding nucleotide DB homology – Mapping a protein to a genome – Mining ESTs and RNA-Seq data for protein similarities
- 9. TBLASTX • TBLASTX – The query is a nucleotide sequence – The database is a nucleotide database – All six frames are translated on the query and on the database • Coding :: Coding homology – Searching distantly-related species – Sensitive but expensive
- 10. O'Reilly Bioinformatics Technology - BLAST Programming 10 BLAST is a heuristic. • A lookup table is made of all the “words” (short subsequences) and “neighboring” words in the query sequence. • The database is scanned for matching words (“hot spots”). • Gapped and un-gapped extensions are initiated from these matches.
- 11. O'Reilly Bioinformatics Technology - BLAST Programming 11
- 12. The Databases (1) • GenBank NR (protein and nucleotide versions) – Non-redundant large databases (compile and remove duplicates) – Anyone can submit, you can call your sequence anything – Low quality; names can be meaningless • Transcriptome Shotgun Assembly (TSA) Database – Transcripts assembled from overlapping ESTs and RNA-Seq reads – Most of the sequences have no annotations
- 13. The Databases (2) • UniProt/Swiss-Prot – Curated from literature – REAL proteins; REAL functions; small; • Genomic databases – Human, Mouse, Drosophila, Arabidopsis, etc. – NCBI, species-specific web pages
- 14. O'Reilly Bioinformatics Technology - BLAST Programming 14 BLAST OUTPUT
- 15. BLAST output 1. List of sequences with scores – Raw score • Higher is better • Depends on aligned length – Expect Value (E-value) • Smaller is better • Independent of length and database size 2. List of alignments
- 16. O'Reilly Bioinformatics Technology - BLAST Programming 16 There are many different BLAST output formats. • Pair-wise report • Query-anchored report • Hit-table • Tax BLAST • Abstract Syntax Notation 1 • XML
- 17. O'Reilly Bioinformatics Technology - BLAST Programming 17 BLAST reports at the NCBI Web page.
- 18. O'Reilly Bioinformatics Technology - BLAST Programming 18 Formatting Page
- 19. O'Reilly Bioinformatics Technology - BLAST Programming 19 Graphical Overview
- 20. O'Reilly Bioinformatics Technology - BLAST Programming 20 One-line descriptions
- 21. O'Reilly Bioinformatics Technology - BLAST Programming 21 Pair-wise alignments
- 22. O'Reilly Bioinformatics Technology - BLAST Programming 22 Query-anchored alignments
- 23. O'Reilly Bioinformatics Technology - BLAST Programming 23 Future improvements: LinkOut, taxonomic and structure links. Link to Locus-link Link to UniGene Link to taxonomy
- 24. O'Reilly Bioinformatics Technology - BLAST Programming 24 BLAST report designed for human readability. • One-line descriptions provide overview designed for human “browsing”. • Redundant information is presented in the report (e.g., one-line descriptions and alignments both contain expect values, scores, descriptions) so a user does not need to move back and forth between sections. • HTML version has lots of links for a user to explore. • It can change as new features/information becomes available.
- 25. O'Reilly Bioinformatics Technology - BLAST Programming 25 Hit-table • Contains no sequence or definition lines, but does contain sequence identifiers, starts/stops (one-offset), percent identity of match as well as expect value etc. • Simple format is ideal for automated tasks such as screening of sequence for contamination or sequence assembly.
- 26. O'Reilly Bioinformatics Technology - BLAST Programming 26 There are drawbacks to parsing the BLAST report and Hit-table. • No way to automatically check for truncated output. • No way to rigorously check for syntax changes in the output.
- 27. O'Reilly Bioinformatics Technology - BLAST Programming 27 Structured output allows automatic and rigorous checks for syntax errors and changes.
- 28. O'Reilly Bioinformatics Technology - BLAST Programming 28 Abstract Syntax Notation 1 (ASN.1) • Is an International Standards Organization (ISO) standard for describing structured data and reliably encoding it. • Used extensively in the telecommunications industry. • Both a binary and a text format. • NCBI data model is written in ASN.1. • Asntool can produce C object loaders from an ASN.1 specification.
- 29. O'Reilly Bioinformatics Technology - BLAST Programming 29 ASN.1 is used for the NCBI BLAST Web page. server ASN.1 Return formatted results BLAST DB Fetch ASN.1 Fetch sequence Request results
- 30. O'Reilly Bioinformatics Technology - BLAST Programming 30 Different reports can be produced from the ASN.1 of one search.
- 31. O'Reilly Bioinformatics Technology - BLAST Programming 31 ASN.1 TaxBlast report Query-anchored BLAST report Pair-wise BLAST report HTML text Hit-table XML HTML text
- 32. O'Reilly Bioinformati 32 The BLAST ASN.1 (“SeqAlign”) contains: • Start, stop, and gap information (zero- offset). • Score, bit-score, expect-value. • Sequence identifiers. • Strand information.
- 33. O'Reilly Bioinformatics Technology - BLAST Programming 33 Three flavors of Seq-Align, Score-block(s) plus one of: • Dense-diag: series of unconnected diagonals. No coordinate “stretching” (e.g., cannot be used for protein- nucl. alignments). Used for ungapped BLASTN/BLASTP. • Dense-seg: describes an alignment containing many segments. No coordinate “stretching”. Used for gapped BLASTN/BLASTP. • Std-seg: a collection of locations. No restriction on stretching of coordinates. Used for gapped/ungapped translating searches. Generic.
- 34. O'Reilly Bioinformatics Technology - BLAST Programming 34 Score Block Score ::= SEQUENCE { id Object-id OPTIONAL , -- identifies Score type value CHOICE { -- actual value real REAL , -- floating point value int INTEGER } } -- integer SEQUENCE is an ordered list of elements, each of which is an ASN.1 type. Required unless DEFAULT or OPTIONAL
- 35. O'Reilly Bioinformatics Technology - BLAST Programming 35 Score Block example 2.45905555x10-9 38.1576692
- 36. O'Reilly Bioinformatics Technology - BLAST Programming 36 Dense-seg definition Dense-seg ::= SEQUENCE { -- for (multiway) global or partial alignments dim INTEGER DEFAULT 2 , -- dimensionality numseg INTEGER , -- number of segments here ids SEQUENCE OF Seq-id , -- sequences in order starts SEQUENCE OF INTEGER , -- start OFFSETS in ids order within segs lens SEQUENCE OF INTEGER , -- lengths in ids order within segs strands SEQUENCE OF Na-strand OPTIONAL , scores SEQUENCE OF Score OPTIONAL } -- score for each seg SEQUENCE OF is an ordered list of the same type of element.
- 37. O'Reilly Bioinformatics Technology - BLAST Programming 37 Dense-seg example
- 38. O'Reilly Bioinformatics Technology - BLAST Programming 38 Std-seg definition Std-seg ::= SEQUENCE { dim INTEGER DEFAULT 2 , -- dimensionality ids SEQUENCE OF Seq-id OPTIONAL , -- sequences identifiers loc SEQUENCE OF Seq-loc , -- locations in ids order scores SET OF Score OPTIONAL } -- score for each segment SET is an unordered list of elements, each of which is an ASN.1 type. Required unless DEFAULT or OPTIONAL. SET OF is an unordered list of the same type of element.
- 39. O'Reilly Bioinformatics Technology - BLAST Programming 39 Std-seg example
- 40. O'Reilly Bioinformatics Technology - BLAST Programming 40 Demo program (“blreplay”) to reproduce BLAST results from ASN.1 • Start/stops and identifiers read in from ASN.1 (SeqAlign). • Sequences and definition lines fetched from BLAST databases.
- 41. O'Reilly Bioinformati 41 Asntool can produce XML from ASN.1 • Really a transliteration, not a new specification • A Document Type Definition (DTD) can also be produced.
- 42. O'Reilly Bioinformatics Technology - BLAST Programming 42 ASN.1 and XML validation differences. • XML can be “well-formed” (does not break any XML syntax rules) or “validated” (checked against a DTD). • ASN.1 must always be valid (checked against a specification).
- 43. O'Reilly Bioinformatics Technology - BLAST Programming 43 Special purpose XML • NCBI specification does not fit the needs of some users (the sequence is not provided in the SeqAlign, when fetched the sequence is packed 2/4 bp’s per byte). • Possible to produce XML with more/less information or in a different format. • First done as an ASN.1 specification, which is then dumped as XML.
- 44. O'Reilly Bioinformati 44 BLAST XML designed to be self- contained. • Query sequence, database sequence, etc. • Sequence definition lines. • Start, stop, etc. (one- offset). • Scores, expect values, % identity etc. • Produced by BLAST binaries and on NCBI Web page.
- 45. O'Reilly Bioinformatics Technology - BLAST Programming 45 Overview of the BLAST XML <!ELEMENT BlastOutput ( BlastOutput_program , BLAST program, e.g., blastp, etc BlastOutput_version , version of BLAST engine (e.g., 2.1.2) BlastOutput_reference , Reference about algorithm BlastOutput_db , Database(s) searched BlastOutput_query-ID , query identifier BlastOutput_query-def , query definition BlastOutput_query-len , query length BlastOutput_query-seq? , query sequence BlastOutput_param , BLAST search parameters BlastOutput_iterations BLAST results for each iteration/run )>
- 46. O'Reilly Bioinformatics Technology - BLAST Programming 46 <!ELEMENT BlastOutput ( BlastOutput_program , BlastOutput_version , BlastOutput_reference , BlastOutput_db , BlastOutput_query-ID , BlastOutput_query-def , BlastOutput_query-len , BlastOutput_query-seq? , BlastOutput_param , BlastOutput_iterations )> <!ELEMENT Iteration ( Iteration_iter-num , Iteration number (one for non PSI-BLAST) Iteration_hits? , Hits (one for each database sequence) Iteration_stat? , Search statistics Iteration_message? Error messages )> <!ELEMENT BlastOutput_iterations ( Iteration+ )>
- 47. O'Reilly Bioinformatics Technology - BLAST Programming 47 <!ELEMENT Iteration ( Iteration_iter-num , Iteration_hits? , Iteration_stat? , Iteration_message? )> <!ELEMENT Hit ( Hit_num , ordinal number of the hit, one-offset (e.g., "1, 2..."). Hit_id , ID of db sequence (e.g., "gi|7297267|gb|AAF52530.1|") Hit_def , definition of the db sequence Hit_accession , accession of the db sequence (e.g., "AAF57408") Hit_len , length of the database sequence Hit_hsps? describes individual alignments )> <!ELEMENT Iteration_hits ( Hit* )>
- 48. O'Reilly Bioinformatics Technology - BLAST Programming 48 <!ELEMENT Hit ( Hit_num , Hit_id , Hit_def , Hit_accession , Hit_len , Hit_hsps? )> <!ELEMENT Hsp ( Hsp_num , ordinal number of the HSP, one-offset Hsp_bit-score , score (in bits) of the HSP Hsp_score , raw score of the HSP Hsp_evalue , expect value of the HSP Hsp_query-from , query offset at alignment start (one-offset) Hsp_query-to , query offset at alignment end (one-offset) Hsp_hit-from , db offset at alignment start (one-offset) Hsp_hit-to , db offset at alignment end (one-offset) Hsp_pattern-from? , start of phi-blast pattern on query (one-offset) Hsp_pattern-to? , end of phi-blast pattern on query (one-offset) Hsp_query-frame? , query frame (if applicable) Hsp_hit-frame? , db frame (if applicable) Hsp_identity? , number of identities in the alignment Hsp_positive? , number of positives in the alignment Hsp_gaps? , number of gaps in the alignment Hsp_density? , score density Hsp_qseq , alignment string for the query Hsp_hseq , alignment string for the database Hsp_midline? )> middle line as normally seen in BLAST report <!ELEMENT Hit_hsps ( Hsp* )>
- 49. O'Reilly Bioinformati 49 Parsing BLAST XML with Expat. • Expat is a popular free-ware used for parsing XML. • Non-validating. • Simple C (demo) program to parse BLAST output.
- 50. O'Reilly Bioinformatics Technology - BLAST Programming 50 Output sizes for a BLASTP search of gi| 178628 vs. nr. • Hit-table: 16 kb • Binary ASN.1 (SeqAlign): 35 kb • Text ASN.1 (SeqAlign): 144 kb • XML (SeqAlign): 392 kb • XML: 288 kb • BLAST report (text): 232 kb • BLAST report (html): 272 kb
- 51. O'Reilly Bioinformatics Technology - BLAST Programming 51 Specification (i.e., “data model”) issues should not be confused with the question about whether to use ASN.1 or XML.
- 52. O'Reilly Bioinformatics Technology - BLAST Programming 52 Structured output is not a panacea. • Design issues must still be addressed. • Semantic issues still exist, e.g. is a start/stop value zero-offset or one-offset. • Data issues still exist, e.g., is the correct sequence shown, are the offsets correct, was the DNA translated with the correct genetic code?
- 53. O'Reilly Bioinformatics Technology - BLAST Programming 53 Overview of BLAST code.
- 54. O'Reilly Bioinformatics Technology - BLAST Programming 54 NCBI toolkit • Has many low-level functions to make it platform independent; supported under LINUX, many flavors of UNIX, NT, and MacOS. • Contains portable types such as Int2, Int4, FloatHi. • Developer should write a “Main” function that is called by a toolkit “main”. • Contains the BLAST code in the “tools” library. • A C++ toolkit is now being developed.
- 55. O'Reilly Bioinformatics Technology - BLAST Programming 55 BLAST code has a modular design. • API for retrieval from databases independent of the compute engine. • Compute engine independent of formatter.
- 56. O'Reilly Bioinformatics Technology - BLAST Programming 56 Readdb API can be used to easily extract information from the BLAST databases. • Date produced. • Title of database. • Number of letters, number of sequences, longest sequence. • Sequence and description of an entry. • Function prototypes in readdb.h.
- 57. O'Reilly Bioinformatics Technology - BLAST Programming 57 Dump a BLAST record in FASTA format (db2fasta.c): Get or display command-line arguments “Main” is called by “main” in the toolkit. Allocate an object for reading the database Get the ordinal number (zero-offset) of the record given a ‘FASTA’identifier (e.g., “gb|AAH06766.1|AAH0676”). Fetch the Bioseq (contains sequence, description, and identifiers) for this record Dump the sequence as FASTA.
- 58. O'Reilly Bioinformatics Technology - BLAST Programming 58 Only a few function calls are needed to perform a BLAST search (doblast.c): Perform a BLAST search of the BioseqPtr query_bsp. The BioseqPtr could have been obtained from the BLAST databases, Entrez or from FASTA using the function call FastaToSeqEntry Allocate a BLASTOptionsBlk with default values for the specified program (e.g., “blastp”), the boolean argument specifies a gapped search Set the expect value cutoff to a non-default value.
- 59. O'Reilly Bioinformatics Technology - BLAST Programming 59 BlastOptionNew BLAST_OptionsBlkPtr BLASTOptionNew (CharPtr progname, Boolean gapped) CharPtr progname: name of program. Legal values are blastp, blastn, blastx, tblastn, and tblastx. Boolean gapped: if TRUE gapped parameters are set, if FALSE ungapped. Non-default values may be specified by changing elements of the allocated structure (typedef in blastdef.h). The most often changed elements (options) are: Nlm_FloatHi expect_value Expect value cutoff Int2 wordsize Number of letters used in making words for lookup table. Int2 penalty Penalty for a mismatch (only BLASTN and MegaBLAST) Int2 reward Reward for a match (only BLASTN and MegaBLAST CharPtr matrix Matrix used for comparison (not BLASTN or MegaBLAST) Int4 gap_open Cost for gap existence Int4 gap_extend Cost to extend a gap one more letter (including first). CharPtr filter_string Filtering options (e.g., “L”, “mL”) Int4 hitlist_size Number of database sequences to save hits for. Int2 number_of_cpus Number of CPU’s to use.
- 60. O'Reilly Bioinformatics Technology - BLAST Programming 60 BioseqBlastEngine SeqAlignPtr BioseqBlastEngine (BioseqPtr bsp, CharPtr progname, CharPtr database, BLAST_OptionsBlkPtr options, ValNodePtr *other_returns, ValNodePtr *error_returns int (LIBCALLBACK *callback) (Int4 done, Int4 positives)) BioseqPtr bsp: contains the query sequence, identifier, and definition line. CharPtr progname: name of program (one of blastp, blastn, blastx, tblastn, or tblastx). CharPtr database: name (and path) to BLAST database(s). Multiple databases to be searched should be separated by a space (e.g., “nt est”). BLAST_OptionsBlkPtr options: BLAST option structure obtained from BLASTOptionNew. If NULL default values will be used. ValNodePtr *other_returns: a linked list of ValNodePtr’s, each one containing information about things like the database(s) searched, the Karlin-Altschul parameters, the region of query masked. See blastall.c to see how to use this information. May be set to NULL. ValNodePtr *error_returns: a linked list of error messages, these may be printed with a call to BlastErrorPrint(error_returns). May be set to NULL. int (LIBCALLBACK *callback) (Int4 done, Int4 positives): callback function to mark progress through the database. May be set to NULL.
- 61. O'Reilly Bioinformatics Technology - BLAST Programming 61 What can I do with the SeqAlignPtr? SeqAlignId gets the (C-structure) identifier for the first (zeroth) sequence. SeqIdWrite formats the information in “query_id” into a FASTA identifier (e.g., “gi|129295”) and places it into query_id_buf. SeqAlignStop returns the end values (zero-offset) for the first and second sequences. SeqAlignStart returns the start value (zero-offset) for the first and second sequences.
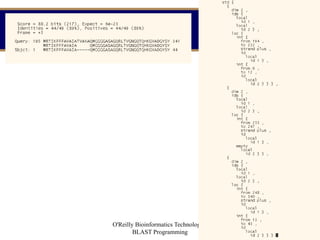
- 62. O'Reilly Bioinformatics Technology - BLAST Programming 62 MySeqAlignPrint output for a search of gi|129295 vs. ecoli
- 63. O'Reilly Bioinformatics Technology - BLAST Programming 63 Notes on Traditional BLAST printing. A call to the fetch function ReadDBBioseqFetchEnable ("blastall", blast_database, db_is_na, TRUE); tells the formatter where to obtain sequences. Entrez or a network connection to the BLAST server could have been used. The one-line descriptions are printed by PrintDefLinesFromSeqAlignEx2(seqalign, 80, outfp, print_options, FIRST_PASS, NULL, number_of_descriptions, NULL, NULL); The pair-wise alignments are printed by ShowTextAlignFromAnnot(seqannot, 60, outfp, NULL, NULL, align_options, txmatrix, mask_loc, FormatScoreFunc); Look at blreplay.c and blastall.c to see details of how these are called.
- 64. O'Reilly Bioinformatics Technology - BLAST Programming 64 Resources • BLAST Home page: http://www.ncbi.nlm.nih.gov/BLAST/ • NCBI Information Engineering Branch home page: http://www.ncbi.nlm.nih.gov/IEB/ • Demonstration programs (parsing XML with EXPAT, blreplay.c, doblast.c, db2fasta.c): ftp://ftp.ncbi.nih.gov/blast/demo
- 65. O'Reilly Bioinformatics Technology - BLAST Programming 65 ASN.1 RESOURCES • The Open Book : A Practical Perspective on OSI by Marshall T. Rose (Prentice Hall). • OSS Nokalva Web site: http://www.oss.com/asn1/overview.html • NCBI toolkit documentation on ASN.1: http://www.ncbi.nlm.nih.gov/IEB/ToolBox/ SDKDOCS/ASNLIB.HTML
- 66. O'Reilly Bioinformatics Technology - BLAST Programming 66 Email addresses • General questions about running BLAST: blast-help@ncbi.nlm.nih.gov • Questions about compiling the toolkit and requests for hard-copy of documentation: toolbox@ncbi.nlm.nih.gov
Editor's Notes
- #3: Similarity is not homology, things may be % similar, but they are either homologous or not. Local aligns active sites on proteins, important since most proteins are modular in nature. A global alignment does not take this into account and similarities may be missed. Statistical theory very important as it tells us whether or not an alignment occurred just by chance.
- #6: Score matrix, e value, parameter
- #10: BLAST is about 100 times faster than exhaustive programs like Smith-Waterman
- #11: Here the word is PQG and neighboring words are everything with a score above 13 (for three letters) as calculated by the given scoring system (e.g., BLOSUM62). PSG is a neighboring word, PQA is not.
- #12: Sequences in the TSA database are at least 200bp in length Ambiguous bases (N’s) should account for less than 10% of the total sequence length
- #16: Most users are familiar with the “Pair-wise” report. The “Query-anchored” report is a variation on that, except the alignments are shown as one-to-many. The Hit-table is a newer format that contains basic start/stop information and is easier to parse. TaxBLAST is a taxonomic centered view of the results. ASN.1 and XML are formal languages and contain structured output. We will see examples of all these formats during the presentation
- #17: Here a protein sequence in FASTA has been entered and the Swiss-prot database selected on the protein-protein (blastp) page.
- #18: This page appears after the last page is sent off and includes a Request Identifier (RID) as well as the results of a Conserved Domain Database (CDD) search. Clicking on “Format!” checks for the results.
- #19: The graphical overview shows the database hits aligned underneath the query sequence (top red bar). Also on this slide is information about the query and the database searched as well as a link to TaxBlast.
- #20: The one-line descriptions consist of four fields: identifier (e.g., gi|116365|sp|P26374|RAE2_HUMAN), a (truncated) definition line, a bit score, and an expect value (false positive rate).
- #21: This is a pair-wise alignment view, part of the query is aligned to one database sequence. In this slide two alignments to the same database sequence are shown. Shown is also the full definition line and the database sequence length as well as some statistics about number of identical letters, etc.
- #22: This is query-anchored. The query sequence is shown aligned with all the database sequences that match to that portion of the query. A dot (“.”) here indicates that the residue is conserved. Note the dinucleotide binding motif from bases 12-22 (Koonin et al., Nature Genetics 12, page 237 (1996)), which consists of bulky hydrophobics and then a glycine rich region. The query-anchored alignments makes it easy to spot others in the database.
- #23: This format is still under discussion. The new version of the BLAST databases contains a flag for GI’s that have entries in Locus-link or UniGene as well as fielded taxonomic information. A flag will also soon be set if a GI has an associated structure that can be viewed with Cn3D and an extra link may be added.
- #24: The BLAST report has been changed or extended for algorithmic changes. PSI-BLAST needed to show on subsequent iterations which hits were new; for gapped BLAST we added the number of gaps in an alignment. We’ve also made changes at user request (e.g., hyper-links for other than first GI in an entry with redundant GI’s).
- #25: The lines starting with “#” should be considered comments and ignored. The last “#” line lists the fields in the Table. The BLAST report contains more information than needed (sequence, definition lines) for most tasks that involves no manual inspection of results.
- #26: The BLAST reports can be large and a full disk can mean truncated output.
- #27: XML would be an example of structured output. For (validating) structured output the specification is an integral step towards building the output. For text reports there is often no specification, but merely an (incomplete) description of the file written afterwards.
- #28: One should not confuse the NCBI data model with ASN.1. The model is written in ASN.1, much as BLAST is written in C or another program is written in FORTRAN. ASN.1 has been in use (very successfully) at the NCBI for 10+ years.
- #29: The ASN.1 we use does not contain the sequence or sequence descriptors, hence they are fetched from the database. What is missing here?…A BLAST search. The search engine and formatter are decoupled so it is possible that one can view a variety of different output formats and only perform one search.
- #30: The “Tax BLAST” report is a taxonomic centered view of the BLAST results.
- #31: The rotating arrow means that both HTML and text can be produced for these reports. XML can also be produced, even though XML is itself a structured and formal language.
- #32: No sequence or sequence descriptions in the SeqAlign, they must be fetched from the BLAST database or some other source.
- #33: We will only discuss Dense-seg and Std-seg in detail, as these are the most often used types.
- #34: The “Object-id” identifies the type of score (e.g., expect value, raw score) and then one (CHOICE) of a REAL or an INTEGER represents the value.
- #35: Here the “score” string identifies the type of score-blk (raw-score) and 19 (an INTEGER) specifies the value.
- #36: “dim” is two here as BLAST always aligns two sequences at a time. It could be larger for a one-to-many alignment. “numseg” is the number of segments in the alignment. The number of segments is actually the 2*(number of gap openings) + 1.
- #37: Here we have three segments, the first starts at (0,0) and has “lens” 14 and is the first third of the alignment. The second segment is the gap in the middle of the alignment; it starts at 14 for the query sequence and has “start” value of –1 for the target sequence to show it is a gap, the “lens” is three. The third segment starts at (17, 14) and has “lens” 16, this is the last third of the alignment.
- #38: There is one Std-seg for each segment of an alignment, so an alignment is really a SEQUENCE OF Std-seg.
- #39: This is from a translating search (blastx). Again three portions (“segments”) to this alignment. The first one has SeqLocs (in the ASN.1) that go from 194-232 for the query (39 bps.) and 0-12 of the target (13 residues). The second segment is the gap and the target SeqLoc shows this by being “empty”. The third segment is from 248-340 (query) and 13-43 (target).
- #40: See last slide for URL to this.
- #44: Here we basically put the BLAST report into XML.
- #45: “?” Indicates zero or one of these (i.e., “BlastOutput_query-seq?”), this would be OPTIONAL in ASN.1.
- #46: The iteration structure is needed for PSI-BLAST. “+” indicates it must occur at least once (e.g., <!ELEMENT BlastOutput_iterations ( Iteration+ )>)
- #47: “*” indicates it may occur more than once (e.g., <!ELEMENT Iteration_hits ( Hit* )>)
- #48: “*” indicates it may occur more than once (e.g., <!ELEMENT Hit_hsps ( Hsp* )>)
- #49: This output shows Query-id, Target definition, expect value, target-id. The point is that you can format the output as you like. See last slide for URL to this demo program. So far only compiled under Solaris.
- #50: The Hit-table does not contain the actual alignment, only the start/stop of an alignment so it’s the smallest. SeqAlign in XML is larger than the SeqAlign in text ASN.1 as XML includes end tags and the tags tend to be larger, in a SeqAlign almost everything is fielded, which is why it’s larger than less well-fielded XML or even the BLAST report.
- #54: Suggest getting the “NCBI Software Development Toolkit”, hard-copy can be obtained by writing to “toolbox@ncbi.nlm.nih.gov”
- #56: We strongly encourage using the readdb API to protect the user against changes in the underlying files. We’re currently putting out a new version of the BLAST databases (though the binaries are back-wards compatible to older databases); the format of the underlying files are subject to change.
- #57: This is written with the NCBI toolkit. See last slide for URL to this demo program. To build this one first needs to compile the NCBI toolkit, then add this to the “makedemo” file.
- #59: These are parameters for the BLAST compute engine, these options have no connection to display (formatting) options. Most of these are self-apparent. The filter_string is really the string you can give to the –F option in blastall or blastpgp. In blastall, blastpgp, and megablast the hitlist_size is set to the maximum of the “number of one-line descriptions” and the “number of database sequences for which alignments are shown”.