Mds&mds mpn
Download as PPT, PDF5 likes2,799 views

This document provides an overview of myelodysplastic syndromes (MDS). MDS are a heterogeneous group of stem cell disorders characterized by cytopenias, dysplastic bone marrow, and risk of leukemia development. The FAB classification from 1982 and revised WHO classification from 2001 are discussed. Key points include defining the subtypes of MDS (such as refractory anemia or RA with excess blasts), associated features (including ring sideroblasts and cytogenetic abnormalities), and diagnostic criteria based on blood and bone marrow findings. In particular, the document highlights the isolated del(5q) abnormality associated with 5q- syndrome.

































































































































































Mds&mds mpn
- 1. Myelodysplastic syndromes MDS/MPN By Azza Mostafa Elkady Assistant lecturer of clinical pathology Assuit Universty A.R.E
- 3. Introduction The myelodysplastic syndromes are a heterogeneous group of clonal stem cell disorders characterized by: • Cytopenias due to impaired blood cell production, • Hypercellular and dysplastic bone marrow, • Increased risk of leukaemic transformation*
- 4. They each exhibit various morphological abnormalities of the blood and bone marrow that are indicative of defective haemopoiesis in one or more lineages.
- 5. Other names • Dysmyelopoietic syndrome • Preleukemic syndrome • Smoldering acute leukemia • Oligoblastic leukemia • Sub acute myelogenous leukemia
- 6. History • The first description of MDS was in 1900 by Leube who used the term leukanamie to describe a patient with severe megaloblastic anaemia that progressed to acute leukaemia. • In the 1930s, the term ‘ refractory anaemia ’ was coined to refer to a group of patients with a macrocytic anaemia that was unresponsive to iron or other dietary haematinics.
- 7. • In the 1950s, it was appreciated that AML in the elderly was often preceded by a pre leukaemic state characterized by peripheral blood cytopenias and increased numbers of blasts in the marrow. • However, the term ‘ pre -leukaemia ’ fell away in the 1970s when it became apparent that many patients never developed leukaemia but died instead of complications arising directly from the cytopenias.
- 8. • In the 1980s, the term ‘ myelodysplasia ’ , or more properly myelodysplastic syndromes to reflect the heterogeneity of the disease, became more widely used.
- 9. Incidence • A disease of the elderly (seventh decade) • Incidence : 3 – 30 /100,000* • Increasing number of therapy related MDS • Males more likely to be diagnosed with MDS than females by a ratio of 1.4 : 1.
- 10. Etiology Primary Unknown …but There is some evidence to suggest that polymorphic variation in certain genes may increase susceptibility to MDS, particularly where the role of the encoded protein* is to counter environmental insults to the cell.
- 11. Secondary – Possible etiologies: Virus, Benzene, cigarette (2 fold risk), Fanconi anemia. – therapy-related (15%) Chemotherapy (alkylating agents)* Radiation Therapy prolonged immunosuppressive therapy autologous transplantation for lymphoma.
- 12. By comparison with primary MDS, these cases of therapy -related MDS are associated with a higher incidence of trilineage dysplasia, genetic abnormalities, evolution to AML and poor response to treatment
- 13. Pathogenesis 1.MDS : a stem cell disorder: • The presence of trilineage dysplasia and cytogenetic abnormalities provides evidence for a multipotent stem/progenitor cell origin. • However, whether this is a haemopoietic stem cell or a myeloid progenitor cell, or a lineage -committed cell in cases of unilineage dysplasia, is not entirely clear.
- 15. • Thus, in 5q – syndrome, the cytogenetic abnormality can be found in both CD34 + CD38 − myeloid progenitors and CD34 + CD19 + pro - B cells, indicative of a true haemopoietic stem cell origin, • while in patients with trisomy 8 the cytogenetic abnormality is often absent from the CD34 + CD38 − fraction suggesting that there might be a different initiating event occurring within the haemopoietic stem cell.
- 16. 2.Immunological a bnormalities in MDS: This is particularly apparent in cases of hypoplastic (10%) MDS that share a number of features in common with aplastic anaemia, • Both appear to be characterized by immune response triggered by abnormal haemopoietic stem cells.
- 17. • Both characterized by a clonal expansion of T cells, Sometimes, the expanded T cells become neoplastic,resulting eventually in the diagnosis of T - cell large granular lymphocytic leukaemia. • clinical presentation with macrocytosis and varying levels of dyserythropoiesis.
- 18. • hypocellular MDS, like aplastic anaemia can respond well to immunosuppressive therapy. Role of immune system in the pathogenesis of MDS supported by the higher incidence of autoimmune disease in these patients.
- 19. 3.Apoptosis in MDS The presence of cytopenias despite a typically hypercellular bone marrow ???? • For those patients undergoing leukaemic transformation,the cytopenias due to maturation block of the malignant cells • However, in cases that lack an excess of blasts, the cytopenias are a reflection of the ineffective haemopoiesis that is a hallmark of the disease.
- 20. The mechanism appears to be one of increased apoptosis of haemopoietic precursors in the marrow. • Apoptosis is more prominent in early MDS, as RA and RARS, than in advanced MDS with excess myeloblasts • This finding is corroborated by flow cytometry analysis of MDS marrow samples to measure relative levels of apoptosis (by annexin V) versus proliferation (by Ki67) that demonstrates a shift from apoptosis to proliferation as the disease progresses.
- 22. FAB classification • In 1982 The FAB group classified MDS according to Morphology and the % of myeloblasts in the BM and PB • These included – Refractory anaemia (RA) – Refractory anaemia with ringed sideroblasts (RARS) – Refractory anaemia with excess blast in marrow (RAEB) – Refractory anaemia with excess blast in transformation (RAEB-t) -CMML
- 23. FAB classification Bone marrow Blood Subtype Dysplasia < 5% blasts As for RA and > 15% Ringed sideroblasts < 1% blasts < 1% blasts Refractory anaemia (RA) Refractory anaemia with ringed sideroblasts (RARS)
- 24. Refractory anaemia with excess blasts < 5% blasts Dysplasia 5– 19% blasts Refractory anaemia with excess blasts in transformation (RAEBt) < 5% blasts Dysplasia 20– 29% blasts or Auer rods Chronic myelomonocytic Leukaemia (CMML) > 1 × 10 9 /L monocytes Dysplasia < 30% blasts
- 26. WHO classification(2001) – Myelodysplastic Syndromes • RA • RARS • RCMD & RCMD-RS • RAEB-1 & RAEB-2 • MDS Unclassified • MDS del(5q) – Myelodysplastic/Myeloproliferative Diseases • CMML • Atypical CML • Juvenile CMML • MDS/MPD, unclassified
- 27. The revised WHO classification of MDS (2008) Subtype Blood Bone marrow 1. Refractory cytopenias with unilineage dysplasia (RCUD) Refractory anaemia (RA) Refractory neutropenia (RN) Refractory thrombocytopenia (RT) Unicytopenia or Bicytopenia No or rare blasts (1%) Dysplasia in > 10% of cells of one myeloid lineage only < 5% blasts < 15% ring sideroblasts
- 28. 2.(RARS) 3.(RCMD) Anaemia No blasts Cytopenia No or rare blasts1% No Auer rods 1 ×10 9 /L monocytes Erythroid dysplasia only >15% ring sideroblasts 5% blasts. Dysplasia in > 10% of cells in two or more myeloid lineages < 5% blasts ± 15% ring sideroblasts
- 29. 4. (RAEB -1) 5.(RAEB -2) Cytopenia(s) < 5% blasts No Auer rods 1 ×10 9 /L monocytes Cytopenia(s) 5– 19% blasts or Auer rods 1 ×10 9 /L monocytes Unilineage or multilineage dysplasia 5– 9% blasts No Auer rods Unilineage or multilineage dysplasia 10– 19% blasts ± Auer rods
- 30. 6.(MDS-U) Cytopenia(s) < 1% blasts Dysplasia in < 10% of cells in one or more myeloid lineage Cytogenetic abnormality supportive of diagnosis < 5% blasts
- 31. 7. MDS associated with isolated del(5q) Anaemia ± thrombo- Cytosis < 1% blasts Prominent megakaryocytes with hypolobated nuclei Isolated del(5q) cytogenetic abnormality No Auer rods
- 32. SUMMARY OF FEATURES OF WHO CATEGORIES Type PB Blasts BM Blasts RS Mono Dysplasia (%) (%) RA 0 <5 <15 <1x109/L E only RARS 0 <5 >15 <1x109/L E only RCMD <5 <5 <15 <1x109/L >10% in >2 lineages RCMD-RS <5 <5 >15 <1x109/L >10% in >2 lineages RAEB 1 <5 5-9 var. <1x109/L E, G, Mega RAEB 2 5-19 10-19 var. <1x109/L E, G, Meg
- 33. MAJOR CHANGES IN WHO CLASSIFICATION OF MDS • Blast count for a diagnosis of AML is reduced from 30% to 20% • RAEB-T is eliminated • Refractory cytopenia with multilineage dysplasia (RCMD) is added • 5q- syndrome is added • CMML is incorporated into a bridging MDS-MPS classification
- 34. 5q- syndrome • Myelodysplastic syndrome associated with isolated del (5q) chromosome • < 5% Blasts in marrow and blood • Predominantly middle-aged to older women • Severe Refractory Anemia (Macrocytic) • Hypercellular marrow with abnormal megakaryocytes.
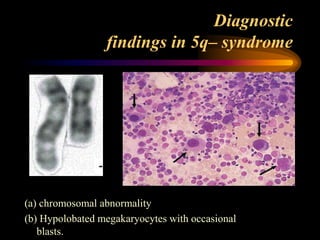
- 35. Diagnostic findings in 5q– syndrome (a) chromosomal abnormality (b) Hypolobated megakaryocytes with occasional blasts.
- 36. 5q- syndrome
- 37. 5q- syndrome
- 38. Myelodysplastic syndrome, unclassifiable Definition: • Is a subtype of MDS which initially lacks findings appropriate for classification into any other MDS category. • Three possible situations which qualify pateints for inclusion in this category.
- 39. 1. Patients with the findings of (RCUD) or (RCMD) but with 1% blasts in the PB qualify for MDS-U. 2.Cases of MDS with Unilineage dysplasia which are associated with pancytopenia. In contrast. the RCUD category only allows for a single cytopenia or bi-cytoperna .
- 40. 3. Patients with persistent cytopenia(s) with 1% or fewer blasts in the blood and fewer than 5% in the BM, unequivocal dysplasia in less than 10% of the cells in one or more myeloid lineages. and who have cytogenetic abnormalities considered as presumptive evidence of MDS.
- 41. • MDS-U patients should be carefully followed for evidence of evolution to a more specific MDS type.
- 42. Diagnosis Clinical features: • Approximately 20% of cases of MDS are detected incidentally • Majority of the remainder present with symptoms and signs of bone marrow failure, (notably fatigue due to anaemia in up to 80% and infections or bleeding in up to 20%).
- 43. • Features of lymphadenopathy, splenomegaly and hepatomegaly are rarely found. • There is association between MDS and several rare disorders that seem to have an immunological basis, including neutrophilic dermatosis (Sweet syndrome), pyoderma gangrenosum and cutaneous vasculitis.
- 44. Lab features: 1. Blood count • Anaemia is the predominant in most patients at presentation, Occurring as pancytopenia in 30 – 50% OR As bicytopenia in 20 – 30%. Isolated neutropenia or thrombocytopenia is rarer, in 5 – 10%. • Occasionally, the blood count is normal and the diagnosis is suggested by abnormal parameters that reflect aberrant morphology, such as RDW that canitself become a useful marker of MDS.
- 45. 2.Peripheral blood morphology RBC While non- specific, but very helpful • Oval macrocytosis (typical) • Reticulocyte count low • Hypochromia (rare; in acquired HbH disease). • Dimorphic cells in sideroblastic anaemia (minority are hypochromic microcytic cells; Pappenheimer bodies by iron stain, basophilic stippling • Megaloblastoid erythroblasts (rare)
- 46. A). Peripheral blood (dimorphic red cells with a population of macrocytes and a population of hypochromic microcytes • http://www.haematologica.org/content/96/6/ 789/F2.large.jpg
- 47. • Peripheral blood from a patient with RAEB, ring sideroblasts and acquired haemoglobin H, showing a grossly dimorphic picture.
- 48. WBC • Hypogranular • Agranular neutrophils highly specific for MDS. • Hypolobated neutrophils (psedo pelger- Huet= Acquired ) classically bilobed ( like spectacles) or even non-lobed (like dumb - bell) pathognomonic.
- 49. Peripheral blood smear with a circulating blast and a pseudo Pelger-Huet cell
- 50. • Monocytosis is present, in CMML and monocytes (often morphologically abnormal. While often reduced in MDS, • basophils and eosinophils might also be raised in the proliferative overlap syndromes. • Type 1( non granular) Type 2 ( granular) blasts may be found in all categories but if in significant numbers are more usually indicative of RAEB.
- 51. Platelets • Hypogranular platelets • Giant platelets
- 52. 3.Bone marrow Aspiration • Bone marrow is hypercellular in majority of patients, • But can be normocellular • in 10 – 20% of cases, hypocellular.
- 53. Dyserythropoiesis • Nuclear budding • Inter-nuclear bridging • Karyorrhexis • Binuclearity & Multinuclearity • Megaloblastoid maturation • Ringed sideroblast (iron staining should performed) • Vacuolation ( in sideroblastic anaemia)
- 58. Perls stain from a patient with RARS showing ring sideroblasts
- 59. Dysgranulopoiesis Quite difficult to appreciate • Nuclear hypolobulation (pseudo-Pelger Heut) • Hypogranularity • Hypersegmentation • prominent basophilic and eosinophilic differentiation • increased numbers of blasts may be present
- 63. Dysmegakaryocytopoiesis • Hypolobulated micro-megakaryocyte • Non-lobulated nuclei in megakaryocyte of all sizes • Multiple, widely separated nuclei
- 65. Megakaryocyte dysplasia with widely separatednuclei
- 69. 4.Bone marrow Biopsy • Assessment of cellularity • Abnormal clustering of megakaryocytes ( micromegakaryocytes) that can be more easily detected by immunohistochemical staining. • BM fibrosis (Reticulin can increased). • ALIP (immature cells present in centre of marrow) - tends to correlate with the blast percentage - may signify propensity to leuk. Trans.
- 70. Abnormal localization of immature precursors
- 71. ALIP
- 72. ALIP stained for neutrophil elastase
- 73. 5.Cytogenetic abnormalities • Cytogenetic analysis of marrow samples plays an important role in the evaluation of MDS with regard to establishing clonality and determining prognosis. • Clonal abnormalities are observed in approximately 50% of primary MDS cases and in up to 90% of cases of secondary TR- MDS
- 74. Cytogenetic abnormalities in MDS Abnormality Primary Therapy - related MDS (%) MDS (%) Complex karyotype 15 – 20 80 – 90 del(5q)/monosomy 5 15– 20 30– 40 del(7q)/monosomy 7 10– 15 40– 50 Trisomy 8 10– 15 10– 15 del(20q) 5– 10 – del(17p) < 5 – del(13q) < 5 – del(11q) < 5 –
- 75. Recurrent chromosomal abnormalities that provide presumptive evidence of primary MDS Abnormality -7 or (7q) -5 or (5q) i(17q) or t(17p) -13 or (13q) (11q) (12p) or t(12p) (9q) idic(X)(q13) t(11;16)(q23;p13.3) t(3;21)(q26.2;q22.1) t(1;3)(p36.3;q21.2) t(2;11)(p21;q23) inv(3)(q21q26.2) t(6;9)(p23;q34) New practical application of 8 color flow cytometry 11-12th Feb 2014
- 76. Monosomy 7 • is the second most common chromosomal abnormality. • lower median age compared with deletions of 5q. • severe refractory cytopenias, and tendency to life - threatening infections. • Monosomy7 confers a poor prognosis
- 77. Cytogenetics and prognosis • Good risk – Normal, isoloted 5q-, isolated 20q-, -Y • Poor risk – Complex changes (> 3 abnormalities) – Chromosome 7 abnormalities • Intermediate risk – All other changes
- 78. 6.Recurrent mutated genes in MDS Gene function Abnormal gene Cell surface receptor KIT , FMS , PDGFRB , GCSFR , MPL , FLT3 Signal transduction NRAS , JAK2 Transcription factor A ML1 , G ATA - 1 , P U.1 , C EBPA ,TP53 Epigenetic factor M LL , A TRX Protein degradation CBL Unknown function TET2
- 79. Known molecular abnormalities in MDS Gene Type of anomaly Incidence (%) RAS (N or K) Point mutation (codon 12, 13 or 61) 10-30% P53 Point mutation or deletion of other allele 5 FMS (encodes M-CSF receptor) Point mutation (codon 969 or rarely 301) 5-10
- 80. 6.Other i nvestigations • Immunophenotyping does not play a major role -low side - scatter, - reduced expression of normal myeloid markers, -and aberrant patterns of expression of other markers. -CD34 expression, and to a lesser degree CD117, often correlates with the blast percentage, while coexpression of CD7 is significant for conferring a worse prognosis.
- 81. Analysis of the (mature) myeloid lineage in normal bone New practical application of 8 color flow cytometry 11-12th Feb 2014
- 82. CD13-CD11b staining of maturing neutrophils New practical application of 8 color flow cytometry 11-12th Feb 2014
- 83. Lineage infidelity markers on immature myeloid cells In approximately 40% of patients: lineage infidelity marker expression detectable on myeloid progenitor cells New practical application of 8 color flow cytometry 11-12th
- 84. Flow Cytometric myeloid dysplasia: myeloid blasts granulocytes (maturing cells) monocytes increased percentage decreased myeloid/lymphoid ratio (<1) decreased/increased number as compared to lymphocytes abnormal granularity* abnormal granularity* abnormal granularity* abnormal expression of CD45 abnormal expression of CD45 abnormal expression of CD45 abnormal expression of CD34 abnormal CD11b/CD13 pattern abnormal expression of CD14 abnormal expression of CD117 abnormal CD16/CD13 pattern abnormal CD11b/HLA-DR pattern abnormal expression of CD13 abnormal expression of CD15 abnormal expression of CD13 abnormal expression of CD33 abnormal expression of CD33 abnormal expression of CD33 abnormal expression of HLA- DR expression of HLA-DR abnormal expression of CD36 expression of CD11b expression of CD34 abnormal expression of HLA-DR expression of CD15 asynchronous shift to the left expression of CD34 expression of lineage infidelity markers CD5, CD7, CD19 or CD56 expression of lineage infidelity markers CD5, CD7, CD19 expression of lineage infidelity markers CD5, CD7, CD19
- 85. Flow Cytometric Scoring System (FCSS) score definition 0 no FC aberrancies in either subpopulation analyzed 1 - a single aberrancy in either granulocytes or monocytes 2 a single aberrancy in both granulocytes and monocytes or … two or three aberrancies in either granulocytes and monocytes or … expression of CD34 or lineage infidelity markers on either granulocytes or monocytes 3 - four or more aberrancies in either granulocytes and monocytes 4 - two or three aberrancies in both granulocytes and monocytes New practical application of 8 color flow cytometry 11-12th
- 86. Flow Cytometric Scoring System (FCSS) score definition + 1 decreased myeloid/lymphoid ratio (<1) normal percentage of myeloid blasts (<5%) with flow cytometric aberrancies + 2 - increased percentage of abnormal myeloid blasts (5-10%) + 3 - increased percentage of abnormal myeloid blasts (11-20%) + 4 - increased percentage of abnormal myeloid blasts (>20%) New practical application of 8 color flow cytometry 11-12th Total flow score: 0-1: normal-mild 2-3: moderate 4-9: severe
- 87. Conclusions: Clinical significance of FC in MDS • FC is instrumental in the diagnosis or exclusion of MDS • Aberrancies on CD34+ cells and myelomonocytic cells by FC is correlated to transfusion dependency and overall survival • FCSS identifies patients at risk for shortened OS within the IPSS-r low risk and good cytogenetic subgroups New practical application of 8 color flow cytometry 11-12th Feb 2014
- 88. • Granulocyte function tests to demonstrate defective phagocytosis, cell killing and motility. • Platelet function tests : reduced aggregation, prolonged bleeding time. • Haemoglobin electrophoresis, or HPLC, to detect HbH (raised in acquired HbH disease) and HbF (raised in juvenile myelomonocytic leukaemia). • Ferrokinetics to assess erythropoiesis → In effective erythropoiesis • Autoantibodies (found in up to 50% of CMML patients). • Serum protein electrophoresis to assess immunoglobulins and detect paraprotein.(↑ polyclonal, ↓ Igs, paraprotein) • Lymphocyte populations to detect altered numbers of T – cell subsets and natural killer cells.
- 89. Diagnostic tool Diagnostic value Priority Peripheral blood smear • Evaluation of dysplasia in one or more cell lines Mandatory • Enumeration of blasts Bone marrow aspirate • Evaluation of dysplasia in one or more myeloid cell lines Mandatory • Enumeration of blasts • Enumeration of ring sideroblasts Bone marrow biopsy • Assessment of cellularity, CD34+ cells, and fibrosis Mandatory Cytogenetic analysis • Detection of acquired clonal chromosomal abnormalities that can allow a conclusive diagnosis and also prognostic assessment Mandatory FISH • Detection of targeted chromosomal abnormalities in interphase nuclei following failure of standard Gbanding Recommended Flow cytometry immunophenotyping • Detection of abnormalities in erythroid, immature myeloid, maturing granulocytes, monocytes, immature and mature lymphoid compartments Recommended SNP-array • Detection of chromosomal defects at a high resolution in combination with metaphase cytogenetics Suggested (likely to become a diagnostic tool in the near future) Mutation analysis of candidate genes* • Detection of somatic mutations that can allow a conclusive diagnosis and also reliable prognostic evaluation Suggested (likely to become a diagnostic tool in Diagnostic approach to MDS 2012
- 90. Minimal diagnostic criteria in MDS A. Prerequisite Criteria • constant cytopenia in one or more cell lineages • exclusion of all other hematopoietic or non- hematopoietic disorders
- 91. B. MDS-related (Decisive) Criteria • dysplasia in > 10% of all cells in one of the lineages, or > 15% ring sideroblasts (iron stain) • 5–19% blast cells in bone marrow smears • typical chromosomal abnormality (karyotyping or FISH)
- 92. C. Co-criteria abnormal phenotype of bone marrow cells by flow cytometry molecular signs of a monoclonal cell population o HUMARA assay, gene profiling, point mutation analysis o markedly and persistently reduced colony- formation (CFU-assay)
- 93. DIFFERENTIAL DIAGNOSIS OF MDS Non-neoplastic causes of myelodysplasia • Megaloblastic anemia • Toxic agents, i.e., heavy metals, acute alcohol intoxication • Drug effects - primarily anti-neoplastic • Congenital dyserythropoietic anemia • Chronic infectious disease • Acquired immunodeficiency syndrome (AIDS) •A.A •NH
- 94. Neoplastic Diseases • Chronic myeloproliferative disease • Acute myeloid leukemia
- 95. Marrow blast percentage: • < 5 0 • 5-10 0.5 • 11-20 1.5 • 21-30 2.0 Cytogentic fetures • Good prognosis 0 (–Y, 5q- , 20q-) • Intermediate prognosis 0.5 (+8, miscellaneous singleabnormality, double abnormalities) • Poor prognosis 1.0 (abnor. 7, complex- >3 abnor.) Cytopenias • None or one type 0 • 2 or 3 type 0.5 Myelodysplastic syndromes IPSS risk-based classification system
- 96. Cytopenias defined as Haemoglobin < 10 g/dL, Neutrophils < 1.8 × 10 9 /L Platelets < 100 × 10 9 /L.
- 99. Emerging new prognostic variables in MDS: 2012 • Bone marrow fibrosis • Co-morbidity score • New karyotyping subgroups (conventional/FISH/flow-FISH) • Newly identified mutations (TET2, EZH2, TP53, RUNX1, NRAS, ASXL1, ETV6, GNAS, SF3B1, and others) • IPSS-revise • Flow cytometric aberrancies (FCSS)
- 100. Manegment
- 102. Myelodysplastic syndrome (MDS) is very uncommen in children, accounting for less than 5% of all haematopoietic neoplasms in patients less than 14 years of age
- 103. • Childhood MDS is recognized as an entity of its own in the current version of the WHO classifi- cation but excludes JMML, which is grouped within the MDS/MPD category. • MDS associated with Down syndrome, which previously accounted for up to 25% of paediatric MDS, is now grouped within a new entity of Down syndrome - related myeloid leukaemia.
- 104. • Many of the morphologic ,immonophenotypic and genetic features observed in MDS in adults are also seen in childhood forms of the disease but there are some significant differences reported
- 105. • For example, the subtypes refractory anaemia with ring sideroblaets and MDS associated with isolated del (5q) chromosomal abnormality are exceedingly rare in children .
- 106. Isolated anaemia, which is the major presenting rnamtestatton of refractory anaemia (RA) in adults, is uncommen in children, who are more likely to present with neutropenia and thrombocytopenia. In addition, hypocellularity of the BM is more commonly observed in childhood MDS than in older patients.
- 107. • In contrast to adult MDS, there are no available studies that have investigated the prognostic significance of distinguishing RAEB-1 and RAEB-2 in children, but it is recommended that this distinction be made for future investigation. • Children with RAEB generally have relatively stable PB counts for weeks or months .
- 108. • The term ‘ refractory cytopenia of childhood ’ is reserved for cases of MDS associated with persistent cytopenia and less than 5% blasts in the marrow and less than 2% blasts in peripheral blood.
- 111. • About 75% of children with RCC show considerable hypocellularity of the bone marrow, making it difficult to differentiate from congenital bone marrow failure syndromes that can lead to secondary myelodysplasia in affected children.
- 112. • These congenital syndromes include disorders such as Fanconi anaemia, dyskeratosis congenita, Schwachman – Diamond syndrome, amegakaryocytic thrombocytopenia, and pancytopenia with radioulnar synostosis.
- 114. Myelodysplastic/ Myeloproliferative Neoplasms • Chronic myelomonocytic leukaemia (CMML) • Atypical chronic myeloid leukaemia (aCML) • Juvenile myelomonocytic leukaemia (JMML) • Myelodysplastic myeloproliferative neoplasm. Unclassifiable (MDS/MPN, U.)
- 115. The WHO classification established a new diagnostic entity for those diseases that share features characteristic of both the myelodysplastic syndromes and the myeloproliferative neoplasms .
- 116. CMML
- 117. CMML • CMML constitutes 20 – 30% of cases of MDS. • The hallmark feature is peripheral blood monocytosis accompanied by morphological dysplasia of other lineages. • It has a male predominance and a median age of presentation of about 70 years (only 10% of CMML cases occur in individuals less than 60 years) • The etiology is largely unknown, therapy - related cases of CMML are very rare.
- 118. Clinical features • Approximately half of patients have splenomegaly, hepatomegaly, at diagnosis. • Individuals with high monocyte counts may develop a maculopapular skin infiltration, gum infiltration, and monocytic pleural and pericardial effusions.
- 119. • Lymphadenopathy is uncommon but when it occurs it may signal a more acute phase with infiltration of lymph nodes by myeloblasts. • Weight loss, fevers and night sweats may occur in symptomatic patients
- 120. laboratory features • The majority of patients present with a leucocytosis. • That is often accompanied by mild anaemia and thrombocytopenia. (Reactive causes due to infection, notably TB, must be excluded) • The monocyte count must be greater than 1 × 1000 /cmm and is usually in the range 2 – 5 × 1000 /cmm, but may exceed 80 × 1000 /cmm.
- 121. • The monocytes generally appear mature but may have agranular cytoplasm and/or abnormal nuclear lobulation (abnormal monocytes)*. • Evidence of dysgranulopoiesis is commonly present, including hypogranular and hypolobated neutrophils that are sometimes difficult to distinguish from dysplastic monocytes
- 122. (A)-WBC is elevated with minimal dysplasia in the neutrophilis. (B)- A normal WBC with absolute monocytosis , nuetropenia and dysgranulopoiesis.
- 123. • Mild basophilia is sometimes present. Eosinophils are usually normal or slightly increased in number, but in some cases eosinophilia may be striking*. • CMML with eosinophilia may be diagnosed when the criteria of CMML are present*. but in addition the eosinophil count in the PB is ≥1500 /cmm.
- 124. • The BM is generally hypercellular, usually with striking granulocytic proliferation and invariably monocytic proliferation that can be distinguished using cytochemical studies. • Typical features of dysplasia can be identified in all three lineages in over 80% of patients
- 125. • The blast cells + promonocytes should account for 5% of peripheral blood leucocytes & and 10% of nucleated marrow cells to give a diagnosis of CMML - 1. • If the blast/promonocyte count is this but 20% in either the PB or BM, or if Auer rods are present, the diagnosis is CMML – 2
- 126. • CMML-2 has worse prognosis and higher risk of transformation to AML, which is diagnosed when the blast/promonocyte count 20%.
- 128. Marrow aspirate from a patient with CMML showing monocytes, promonocytes and blast cells.
- 129. The folded nucluei and delicate nuclear chromatin can be appreciated among granulocytes.
- 131. • Lysozyme used in conjunction with cyto- chemistry for CAE can also facilate the identifica tion of monocytic cells,which are lysozyme-positive but negative for CAE, in contrast with the granulocytrc precursor cells, which are positive for both.
- 132. The monocytic component stained with naphthol-ASD- chloroacetate estrase raection combined with alpha naphthyl butytrate estrase (monocyte brown, nutrophil blue, dual staining cells have mixture of brown & blue.
- 133. • Imunohistochemistry on tissue sections for the identification of monocytic cells is relatively insensitive as compared with cytochemistry or flow cytometry. • The most reliable markes ars are CD 68 and CD l63
- 134. BM biobsy section stained with CD 163, note the postivity of the scattered monocytic cells as well as the strong positivity in the macrophage in the stain.
- 135. Immunophenotyping may be helpful for identifying myelomonocytic populations • The PB and BM cells usually express CD33 and CD13, with variable expressionof CD14, CD68 and CD64. • but can also give prognostic information such as reduced CD14 expression indicating monocytic immaturity, aberrant expression of CD2 and CD56, and the CD34 positive cell percentage.
- 136. Cytogenetic analysis is important for confirming clonality, although abnormalities are only found in 30 – 40% of cases,
- 137. • Up to 40% of patients have point mutations of RAS genes. • Hypermethylation of the CDKN2B gene (which encodes the tumour suppressor p15INK4b), resulting in reduced expression,in about 50% of patients.. • Recently, the CBL gene, has been implicated in progressive CMML
- 138. • Exclude certain chromosomal translocations, by cytogenetic and (PCR), that are indicative of alternative diagnoses. 1. BCR – ABL1 rearrangement (CML). 2.(eosinophilia - related disorders) associated with abnormalities of PDGFRA , PDGFRB and FGFR1. 3. Mutations of the JAK2 gene
- 139. WHO diagnostic craitria of CMML
- 141. • Atypical chronic myeloid leukaemia is a poorly defined entity. • Importantly, it is negative for the BCR – ABL1 fusion gene of typical chronic myeloid leukaemia • aCML principally involving the neutrophil lineage.
- 142. • Myeloid precursors often comprise more than 10% of cells but blasts are rarely more than 5%. • Dysgranulopoiesis is prominent but multilineage dysplasia is also seen.
- 143. • Patients usually present with anaemia and/or thrombocytopenia that accompany the leucocytosis. • Splenomegaly is common and often causes symptoms because of its massive enlargement.
- 145. • Cytogenetic abnormalities can be identified in 80% of cases although is none specific. • The outlook is generally poorer for atypical chronic myeloid leukaemia than for CMML, with a median survival of less than 20 months. .
- 147. JMML
- 148. • JMML is a clonal haemopoietic disorder of childhood characterized principally by proliferation of the granulocytic and mono cytic lineages. • comprising approximately 2 – 3% of all childhood leukaemias but 40% of childhood MDS.
- 149. • The majority of cases occur in children under 3 years and twice as commonly in boys than girls. • There are associations with neurofi bromatosis type 1 and Noonan syndrome due to germline mutations in the NF1 and PTPN11 or KRAS genes respectively
- 150. • There is marked in vitro hypersensitivity of myeloid progenitors to (granulocyte/macrophage colony - stimulating factor) that is a hallmark feature of JMML and suggestive of defective RAS – MAP kinase signalling that is often attributable to RAS mutations. • Monosomy 7 is the most common chromosomal abnormality, found in 25% of cases.
- 151. • Clinically, most patients present with constitutional symptoms or evidence of infection • and are found to have marked hepatosplenomegaly • Lymphoid and tonsillar enlargement is also common.
- 152. • Typically, there is a leucocytosis comprising neutrophils, • myeloid precursors and monocytes, with blasts constituting less than 5% of cells. The marrow is hypercellular and dysplastic features are minimal.
- 154. • A marked increase in the synthesis of HbF is a recurrent finding that has poor prognostic implications. • The prognosis of JMML is variable with a median survival of 1 year. It is usually rapidly fatal without treatment, causing organ failure, especially respiratory failure due to leukaemic infiltration, while blast transformation occurs infrequently.
- 156. MDS/MPN, U.
- 158. Refractory anaemia with ring sideroblasts and thrombocytosis
- 159. • The precise nature of (RARS - T) is a controversial and unresolved issue. • These patients meet the criteria for RARS but also have persistently elevated platelet counts over 450 × 10 9 /L. • The majority (50 – 60% of cases) carry the V617F mutation of the JAK2 gene that is more commonly associated with myeloproliferative disorders.
- 160. • Whether RARS - T represents a JAK2 - positive myeloproliferative neoplasm with acquired dysplastic features or, • Conversely, a form of MDS with an acquired proliferative mutation remains a question ? • For this reason, RARS - T exists as a provisional entity in the current version of the WHO classification.
Editor's Notes
- #4: approximately 40% of patients will transform to AML
- #10: MDS in adults is 3 – 4 per 100 000. However, this rate increases markedly with age, exceeding 30 per 100 000 for individuals over the age of 80 years.
- #11: enzymes for the metabolism of carcinogens, proteins involved with oxidative stress,repair damaged DNA.
- #12: alkylating agents,as cyclophosphamide, and topoisomerase II inhibitors,as etoposide, &antimetabolite drugs
- #121: however, the WBCs normal or slightly decreased with variable neutropenia and the disease resembles MDS., nearly one half of patients it is increased due not only to monocytosis but also to neutrophilia
- #122: But in comparison to promonocytes (and monoblasts). Have denser chromatin, nuclear convolutions and folds, and a more greyish cytoplasm
- #124: Patients in this category may have complications related to the degranulation the eosinophils. These &quot;hypereosinophilic &quot; cases of CMML may
- #140: Blasts include myeloblasts, monoblasts and promonoblast.