





















SEB submissions in Canada
- 1. Presented by Neha Gandhi Yogender Arya 1
- 2. Bio-therapeutic products also known as biologic drugs have a successful record in treating many life-threatening and chronic diseases. The recent expiration of patents and/or data protection for the first major group of originator biologics and the anticipation of additional expirations has led to the development of products that are designed to be “similar” to a licensed originator product. In Canada, they are referred to as Subsequent Entry Biologics (SEBs), copies of previously approved biological products. By Yogendra Arya and Neha Gandhi 2
- 3. Biologics SEBs Biologics are drug products derived SEB is defined by Health Canada as from biological sources and include “a biologic drug that enters the gene therapies, vaccines, antibodies market subsequent to a version and other therapeutic products previously authorized in Canada, and derived through biotechnology with demonstrated similarity to a reference biologic drug. In order for a product to be considered an SEB it should have the same therapeutic effects as the originally approved biologic, a similar safety profile and the same guarantees to be free of contaminating viruses or other infectious agents. SEB manufacturers may make reference to the information contained in an innovator’s biologic drug submission – that is, the reference biologic drug submission – in order to reduce the amount of clinical data required to obtain marketing approval for its product. By Yogendra Arya and Neha Gandhi 3
- 4. The Biologics and Genetic Therapies Directorate (BGTD) regulates biologics under Divisions 1, 1A, 2, 4, 5, and 8 of Part C of the Food and Drug Regulations. Information provided by manufacturers is evaluated to verify that the biologic meets quality, safety and efficacy requirements, and that the benefits of the biologic outweigh the risks for its intended use. A Notice of Compliance (NOC) and Drug Identification Number (DIN) are issued to sponsors when their products are authorized for sale (distribution) in Canada. After a biologic has received an NOC, the manufacturer must adhere to BGTD's Lot Release Program, which sets out risk-based criteria for testing of the product prior to its release onto the Canadian market. By Yogendra Arya and Neha Gandhi 4
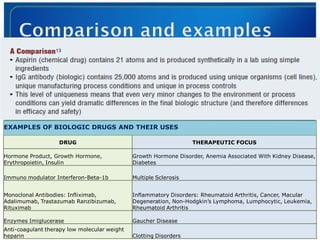
- 5. EXAMPLES OF BIOLOGIC DRUGS AND THEIR USES DRUG THERAPEUTIC FOCUS Hormone Product, Growth Hormone, Growth Hormone Disorder, Anemia Associated With Kidney Disease, Erythropoietin, Insulin Diabetes Immuno modulator Interferon-Beta-1b Multiple Sclerosis Monoclonal Antibodies: Infliximab, Inflammatory Disorders: Rheumatoid Arthritis, Cancer, Macular Adalimumab, Trastazumab Ranzibizumab, Degeneration, Non-Hodgkin’s Lymphoma, Lumphocytic, Leukemia, Rituximab Rheumatoid Arthritis Enzymes Imiglucerase Gaucher Disease Anti-coagulant therapy low molecular weight By Yogendra Arya and Neha Gandhi 5 heparin Clotting Disorders
- 6. The concept of an SEB applies to all biologic drug submissions where the sponsor seeks authorization for sale based on demonstrated similarity to a previously approved biologic drug. Existence of Reference Biological Drug authorized for sale sufficient efficacy and safety data The product (i.e., SEB) can be well characterized by a set of modern analytical methods The SEB, through extensive characterization and analysis, can be judged similar to the reference biologic drug by meeting an appropriate set of pre- determined criteria. It applies to biologic drugs that contain, as their active substances, well characterized proteins derived through modern biotechnological methods such as use of recombinant DNA and/or cell culture. By Yogendra Arya and Neha Gandhi 6
- 7. The responsibility of sponsor to provide the necessary evidence to support the application The Food and Drugs Act and Regulations within the existing regulatory frameworks for biologic, pharmaceutical, and generic pharmaceutical drugs Similarity between the SEB and Reference Authorization of an SEB is not a declaration of pharmaceutical or therapeutic equivalence to the reference biologic drug. All SEBs are subject to the laws, and patent and intellectual property principles outlined within the Food and Drug Regulations (Data Protection), Patented Medicines (Notice of Compliance) Regulations, and the Patent Act. Once a Notice of Compliance (NOC) is issued, the SEB is a new biologic drug and regulated accordingly. However, an SEB should not be used as a reference biologic drug for another SEB submission. By Yogendra Arya and Neha Gandhi 7
- 8. Applicable Regulations: Subject to the Food and Drug Regulations for authorization and oversight for the following: C.08.002 (1)(a): No person shall sell or advertise a new drug unless the manufacturer has filed with the Minister a New Drug Submission (NDS) relating to the new drug that is satisfactory to the Minister. C.08.002 (2): A New Drug Submission shall contain sufficient information and material to enable the Minister to assess the safety and efficacy of a new drug. By Yogendra Arya and Neha Gandhi 8
- 9. Reference Biologic Drug Suitable to support the submission; Authorized for sale and should be marketed in Canada; The same reference biologic drug should be used throughout the studies supporting the safety, quality, and efficacy of the product Same dosage form, strength, and route of administration The active substance must be shown similar; An SEB should not be used as a reference biologic drug; and Should have adequate safety, efficacy, and effectiveness data By Yogendra Arya and Neha Gandhi 9
- 10. Regulated by Part C, Division 8 of the Food and Drug. Submission requirements for SEBs will be determined on a case by case basis, and may include, but not be limited to: A complete chemistry and manufacturing data package A rationale for the choice of the innovator biologic and extensive published information on its safety and efficacy. Sufficient characterization information to demonstrate both chemical and biological comparability to the innovator product. Sufficient comparative animal toxicity and toxicological data, where appropriate. By Yogendra Arya and Neha Gandhi 10
- 11. Pharmacodynamic data to demonstrate comparable bioactivity based on parameters or surrogate markers that are clinically relevant and validated. Pharmacokinetic data to demonstrate comparable bioavailability of the SEB to the innovator product based on suitable validated pharmacokinetic parameters. Data characterizing the immunogenic profile of the SEB in humans and its potential impact on safety and efficacy. A clinical package which demonstrates the safety and efficacy of the SEB including comparative studies between the SEB and innovator products By Yogendra Arya and Neha Gandhi 11
- 12. Clinical trials conducted in Canada involving SEBs are subject to Part C, Division 5 of the Food and Drug Regulations, which outlines the requirements applicable to the sale and importation of drugs for use in human clinical trials in Canada. Sponsors need to include all information identified in C.05.005 of the Food and Drug Regulations in their application for authorization. If a non-Canadian reference biologic drug is used in clinical studies in Canada, data must be provided to support its safety and to satisfy the intent of the regulatory requirements for chemistry and manufacturing information. . By Yogendra Arya and Neha Gandhi 12
- 13. The target time for review of an SEB will be the same as that for an NDS. Sponsors of SEBs are encouraged to consult with BGTD for regulatory guidance at any stage of the development of an SEB. By Yogendra Arya and Neha Gandhi 13
- 14. In addition to a full chemistry and manufacturing (C&M) data package that is expected for a standard new biologic drug, the SEB package should provide extensive data on the demonstration of similarity with the reference biologic drug, including characterization Studies conducted in a side-by-side format. By Yogendra Arya and Neha Gandhi 14
- 15. The assessment of similarity should be organized in the Common Technical Document (CTD) as a distinct collection of data in module 3 with an associated section in the Quality Overall Summary containing appropriate cross-references. However, the reorganization of modules 2 and 3 of a CTD submission already prepared for another regulatory jurisdiction should not be necessary By Yogendra Arya and Neha Gandhi 15
- 16. Once granted an NOC, an SEB is considered to be a new (“stand-alone”) product with all of the associated regulatory requirements. For any changes to the manufacturing process that warrant a demonstration of comparability, the products to be compared will be the pre-change and post-change versions of the SEB. Comparisons with the original reference biologic drug are not required. By Yogendra Arya and Neha Gandhi 16
- 17. An SEB product sponsor is eligible to apply for one or more clinical indications granted to the reference biologic drug in Canada. Where a clinical indication being sought is not held by the reference biological drug, full clinical trial data shall be provided in support of that indication. Non-clinical studies: In vitro studies In vivo studies Clinical studies: Pharmacokinetic studies Pharmacodynamic studies Clinical efficacy and safety trials By Yogendra Arya and Neha Gandhi 17
- 18. An SEB is not the first in its class to be brought to market; it is therefore important that an RMP be presented prior to issuance of marketing authorization. The RMP should be designed to monitor and detect both known inherent safety concerns and potentially unknown safety signals that may result from the impurity profile and other characteristics of the SEB. By Yogendra Arya and Neha Gandhi 18
- 19. A PvP should be provided and should include the submission of periodic safety update reports (PSURs). The PSURs for an SEB should include a discussion of the benefit-risk balance of the SEB post-market. By Yogendra Arya and Neha Gandhi 19
- 20. ADR reporting is required post-market under section C.01.016 of the Food and Drug Regulations: Any serious ADR that is reported requires the manufacturer of that drug to submit all information with respect to that report within 15 days after receiving the information. Furthermore, on an annual basis or as requested by the Director, the manufacturer will conduct a concise, critical analysis of the adverse drug reactions and serious adverse drug reactions after such an occurrence. After an analysis, the Director may request written summary reports where safety is questionable. By Yogendra Arya and Neha Gandhi 20
- 21. Unlike generic pharmaceutical drugs, the sponsor of an SEB will not be able to utilize the PM of the reference biologic drug in its entirety as that of its own product. The contents of the PM for SEBs will include following information: A statement indicating that the product is an SEB Key data on which the decision for market authorization was made Tables showing the results of the comparisons between the SEB and reference biologic Drug Information on the indications approved for use There should be no claims for bioequivalence between the SEB and reference biologic Drug There should be no claims for clinical equivalence between the SEB and the reference biologic drug By Yogendra Arya and Neha Gandhi 21
- 22. It is Health Canada’s intention to harmonize as much as possible with other competent regulators and international organizations such as the World Health Organization. Hence, Health Canada may be adapting suitable definitions, terminology, and applicable guidance documents. Health Canada recommends that sponsors refer to the product class specific guidance documents developed by the Similar Biological (Biosimilar) Medicinal Products Working Party, European Medicines Agency as the scientific principles are consistent with those of Health Canada. By Yogendra Arya and Neha Gandhi 22
- 23. Guidance for Sponsors: Information and Submission Requirements for Subsequent Entry Biologics (SEBs) http://www.hc-sc.gc.ca/dhp mps/alt_formats/pdf/brgtherap/ applic-demande/guides/seb-pbu/seb-pbu-2010-eng.pdf By Yogendra Arya and Neha Gandhi 23