SINDROME DE DOWN
Descargar como PPTX, PDF6 recomendaciones5,551 vistas

El documento define el Síndrome de Down como una condición genética causada por la presencia de un cromosoma extra 21. Describe los signos físicos comunes como rasgos faciales distintivos, pliegues en las manos y retraso en el desarrollo. Explica que puede diagnosticarse antes o después del nacimiento a través de pruebas genéticas y exámenes físicos. El tratamiento se enfoca en terapias para mejorar habilidades y el manejo de cualquier problema médico asociado.











































SINDROME DE DOWN
- 1. SINDROME DE DOWN. Por: Diana C. Moreno, Yilena M. Ordoñez y Juan F. Paz, Andrés Muñoz, Andrés Erazo, Diego Villafañe, Christian Molina, Alejandra Ospina, Mónica Rodríguez y Antonio Medina
- 2. DEFINICION El Síndrome de Down es una de las causas genéticas más comunes de retraso mental o de desarrollo. Es un trastorno genético en el cual una persona tiene 47 cromosomas en lugar de los 46 usuales. Los síntomas del síndrome de Down varían de una persona a otra y pueden ir de leves a graves. Sin embargo, los niños con síndrome de Down tienen una apariencia característica ampliamente reconocida.
- 3. LOS SIGNOS FISICOS ABARCAN: Disminución del tono muscular al nacer Exceso de piel en la nuca Nariz achatada Uniones separadas entre los huesos del cráneo (suturas) Pliegue único en la palma de la mano Orejas pequeñas Boca pequeña Ojos inclinados hacia arriba Manos cortas y anchas con dedos cortos Manchas blancas en la parte coloreada del ojo (manchas de Brushfield)
- 4. Los bebés con Síndrome de Down: - Tienden a desarrollarse más lentamente que otros bebés - Empiezan a caminar más tarde. - Cuando crecen, tienden a ser más lentos y retraídos que los otros miembros de la familia - Pueden ser más bien robustos o de constitución ancha. - En muchos casos, tienen los párpados ligeramente hacia arriba. Podrían tener pequeños pliegues de piel en el rabillo interior de los ojos. Sus narices pueden ser un poco aplanadas y las orejas pueden ser pequeñas y con forma anormal.
- 5. FACTORES DE RIESGO EXTRINSECOS. RADIACIONES(Rayos X) VIRUS AGENTES QUIMICOS MUTAGENOS FACTORES INMUNOBIOLOGICOS
- 6. FACTORES DE RIESGO INTRINSECOS - FACTORES HEREDITARI OS. - OTROS FACTORES (Edad de la madre).
- 8. TROB 21Q/21Q TROB 14Q/21Q TRASLOCACIÓN ROBERTSONIANA Down: • 3 copias completas del cromosoma 21 • 3 copias del brazo largo del cromosoma 21
- 9. Translocación Translocación robertsoniana Intercambio de material entre dos o más cromosomas, existen dos tipos: reciproca y robertsoniana. Ruptura de los brazos cortos de los cromosomas acrocéntricos, seguida de la fusión de las porciones largas Para formar un solo cromosoma. 46,XY,trob (14q/21q) 46,XY,trob (21q/21q)
- 11. (21) (21) 46 CROMOSOMAS 45 CROMOSOMAS 45,XX,trob (21q/21q) EXPOSICIONES 45 CR 44 Cr 21/21 23 CROM. 21/21 46 CROM. 21/21 21 Inviables: monosomias del crom 21 22 cr22 cr22 cr 100% de posibilidades de descendencia con down 46,XY,trob (21q/21q) trisomia universal
- 13. NO DISYUNCION MITOTICA 46 4646 46 46 45 47 • SE PRODUCEN 3 LINEAS CELULARES: 45/46/47. • MOSAICO: (46,XX / 47,XX +21). • EL RESULTADO FINAL ES UN PRODUCTO CON DOS POBLACIONES DE CÉLULAS: NORMALES Y TRISÓMICAS, ES DECIR, UN MOSAICO CELULAR. • EL CUADRO FENOTÍPICO ES VARIABLE SEGÚN SEA LA PROPORCIÓN DE CÉLULAS NORMALES Y TRISÓMICAS. cigoto Mitosis Mitosis No separación
- 14. 46 4747 47 cigoto No separación Mitosis Mitosis • SE PRODUCEN 2 LINEAS CELULARES: 45/47. • UNIVERSAL: (47,XX+21). • SE PRODUCE EN LA PRIMERA DIVISIÓN CELULAR POR FALTA DE DISYUNCIÓN DE LA CÉLULA DE ORIGEN. • UNA CÉLULA RECIBE ENTONCES 3 CROMOSOMAS 21 Y LA OTRA RECIBE 1 CROMOSOMA 21. • EL EMBRIÓN SE DESARROLLA ENTONCES DE MODO QUE TODAS SUS CÉLULAS TIENEN 3 CROMOSOMAS 21.
- 15. NO DISYUNCION MEIOTICA 46 24 24 24 22 22 22 ovogonia Meiosis II • NO SEPARACIÓN EN LA MEIOSIS I. • ES EL CASO MÁS FRECUENTE, ABARCA EL 90 A 95 %. ESTA ANOMALÍA SE HALLA PRESENTE ANTES DE LA FERTILIZACIÓN. • PUEDE OCURRIR EN LA ENFERMEDAD QUE EXISTA UNA MALA DISTRIBUCIÓN ERRÓNEA DE LOS CROMOSOMAS EN LA FORMACIÓN DEL ÓVULO O DEL ESPERMATOZOIDE, EN LOS CUALES UNO TENDRÁ 2 CROMOSOMAS 21. • DESPUÉS DE LA CONCEPCIÓN, EL CIGOTO FERTILIZADO CONTIENE 3 CROMOSOMAS 21. • TODAS LAS CÉLULAS DEL CUERPO TENDRAN 3 CROMOSOMAS 21.
- 16. 46 2323 2323 24 22 ovogonia Meiosis II • NO SEPARACIÓN EN LA MEIOSIS II. • SE PRODUCIRA UN MOSAISISMO YA QUE SE PUEDE OBSERVAR DE QUE POSEE UNA LINEA DE CELULAS NORMALES.
- 17. INCIDENCIA GLOBAL ENTRE 1 / 1000 Y 1/1100 nacimientos 15-29 años 1/1,500 nacidos vivos 30-34 años 1/ 800 nacidos vivos 35-39 años 1/385 nacidos vivos 40-44 años 1/106 nacidos vivos 45 + 1/30 nacidos vivos EDAD MATERNA EXPECTATIVA DE VIDA: 50 AÑOS APROX
- 18. DIAGNOSTICO El diagnostico del síndrome de Down puede hacerse antes del parto o tras él. En este último caso se hace con los datos que proporciona la exploración clínica y se confirma posteriormente mediante el cariotipo. Las pruebas prenatales pueden ser de sospecha (screening) o de confirmación. Estas últimas se suelen realizar únicamente si existen antecedentes de alteraciones genéticas, si la mujer sobrepasa los 35 años o si las pruebas de screening dan un riesgo alto de que el feto presente síndrome de Down.
- 19. PRUEBAS DE PRESUNCIÓN En el primer trimestre del embarazo: se dispone del examen ecográfico del cuello del feto(translucencia nucal) donde se nota un engrosamiento de la nuca. Segundo trimestre: se pueden emplear exámenes ecográficos, donde se observa el tamaño de los huesos o de órganos.
- 20. PRUEBAS INVASIVAS Se realizan cuando las pruebas de presunción arrojan un valor positivo a) Amniocentesis Primero se identifican y localizan la placenta y la cavidad amniótica mediante ecografía, y se administra un anestésico local. Después, y bajo control directo ecográfico, se inserta una aguja en el vientre a través de la pared abdominal y se aspira el líquido amniótico que drena por la aguja. Posteriormente se centrifuga este líquido y las células fetales que se obtienen se dejan crecer en cultivo para hacer después el análisis cromosómico.
- 21. b) Biopsia de las vellosidades corionicas. La prueba consiste en la obtención de una pieza de tejido placentario por vía vaginal o a través del abdomen, generalmente entre la 8ª y la 11ª semana de gestación a )
- 22. c) Cordocentesis Es un método excepcional que consiste en la punción del cordón umbilical a través de la pared del vientre de la madre para obtener sangre fetal directa. Su riesgo de pérdida fetal es mayor que los anteriores métodos (3%).
- 23. CUADRO CLINICO
- 24. Disminución del tono muscular en forma generalizada o focal, que generalmente se asocia a déficit en el desarrollo psicomotor. Suele ser ancha y rectangular; el dorso se presenta aplanado debido a una escasa formación de los huesos nasales. (puente nasal deprimido)
- 25. Cuello: suele ser corto y ancho. Cráneo: es pequeño, su parte posterior está ligeramente achatada, algunos presentan áreas donde falta el cabello presentan un pliegue de la piel en la esquina interna de los ojos (llamado epicanto).
- 26. LENGUA: Debido a la falta de tono muscular tiene tendencia a salirse de la boca. BOCA: la boca se mantiene abierta Suele ser irregular e incompleta (anodoncia), la forma de los dientes es a veces anómala y tiene alteraciones en el esmalte.
- 27. En las palmas de las manos muestran un único pliegue transversal, con dedos cortos que se curvan hacia adentro. Dermatoglifos atípicos
- 28. Generalmente se seca y se agrieta con facilidad. la laringe parece estar situada más allá de lo habitual por lo tanto la voz es gutural y su articulación difícil.
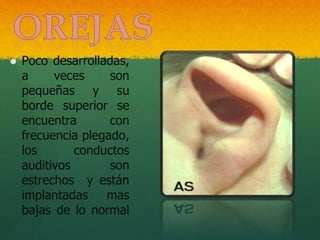
- 29. Poco desarrolladas, a veces son pequeñas y su borde superior se encuentra con frecuencia plegado, los conductos auditivos son estrechos y están implantadas mas bajas de lo normal
- 30. Características Porcentaje de aparición Características Porcentaje de aparición Retraso mental 100% Microdoncia total o parcial 60% Retraso del crecimiento 100% Puente nasal deprimido 60% Dermatoglifos atípicos 90% Clinodactilia del 5º dedo 52% Diástasis de músculos abdominales 80% Hernia umbilical 51% Hiperlaxitud ligamentosa 80% Cuello corto 50% Hipotonía 80% Manos cortas/braquidactilia 50% Braquiocefalia/región occipital plana 75% Cardiopatía congénita 45% Genitales hipotróficos 75% Pliegue palmar transversal 45% Hendidura palpebral 75% Macroglosia 43% Extremidades cortas 70% Pliegue epicántico 42% Paladar ojival 69% Estrabismo 40% Oreja redonda de implantación baja 60% Manchas de Brushfield (iris) 35%
- 32. No se conocen con exactitud las causas que provocan el exceso cromosómico, aunque se relaciona estadísticamente con una edad materna superior a los 35 años. Las personas con síndrome de Down tienen una probabilidad algo superior a la de la población general de padecer algunas enfermedades, especialmente de corazón, sistema digestivo y sistema endocrino, debido al exceso de proteínas sintetizadas por el cromosoma de más.
- 33. Tratamiento • No se puede curar. Sin embargo, el tratamiento temprano puede ayudar a muchas personas con síndrome de Down para vivir una vida productiva en la edad adulta. • Se pueden beneficiar de la terapia de habla, terapia ocupacional y ejercicios para ayudar a mejorar sus habilidades motoras. También podrían ser ayudados por la educación especial y la atención en la escuela.
- 34. T R A T A M I E N T O Algunos de los problemas médicas comunes en las personas con síndrome de Down, como cataratas, problemas de audición, problemas de tiroides y convulsivos, también se puede tratar o corregir. Se ha sugerido que los niños con síndrome de Down pueden beneficiarse de un tratamiento médico que incluye los suplementos de aminoácidos y un fármaco conocido como Piracetam que puede mejorar la capacidad del cerebro para aprender y entender.
- 35. Acompañamiento genético. El 95% aproximadamente de los S. de Down se deben a una trisomía 21 regular. Esta trisomía 21 se produce por una no-disyunción en la célula germinal materna, es decir en el óvulo o por una no disyunción en la primera división mitótica. Esto explica que en más del 90% de los casos el cromosoma 21 extra en el S. de Down sea de procedencia materna.
- 36. Aproximadamente el 4% de los S. de Down se deben translocaciones ya sean heredadas o "de novo".
- 37. ¿Qué riesgo existe de que una pareja tenga un hijo/a Down? La materno-edad-dependencia repetidamente demostrada en el S. de Down pone de manifiesto que cuanto más elevada es la edad de la madre, mayor es el riesgo de tener un hijo afecto de S. de Down , hecho que también sucede con el resto de las trisomías autosómicas. La asociación entre edad materna y no-disyunción se ha discutido y hay autores que sostienen que a medida que aumenta la edad de la madre aumenta el riesgo de no-disyunción.
- 38. Relación con la edad: Otros estudios sugieren que las madres más jóvenes que quedan embarazadas de un embrión afecto de trisomía 21 tienen mayor tendencia al aborto espontáneo mientras que las más añosas "toleran" mejor estos embriones trisómicos, lo que explicaría la materno-edad-dependencia del Síndrome de Down. Conviene señalar que a pesar de lo anteriormente expuesto el 80% de los niños downianos nacen de madres que tienen menos de 35 años, lo cual no contradice la materno-edad-dependencia, ya que la fertilidad es mayor en las madres jóvenes.
- 39. El riesgo de recurrencia de tener otro hijo con S. de Down, va a depender del tipo de S. de Down que tiene el hijo afecto. Cuando en una pareja con cariotipo normal se produce el nacimiento de un hijo con trisomía 21 regular, el riesgo de recurrencia es bajo, aproximadamente del 1%-2% pero sin duda más elevado que el de una pareja de la misma edad. En caso de translocación "de novo", con cariotipo obviamente normal de los padres, el riesgo de recurrencia de tener otro hijo con S. de Down por translocación "de novo" es muy bajo, inferior al 1%. ¿Qué riesgo existe de que una pareja que ya ha tenido un hijo con S. de Down, tenga otro con la misma enfermedad?
- 40. En los casos de translocación heredada el riesgo de recurrencia dependerá de dos factores: 1) del tipo de translocación 2) de quien es el portador de la translocación equilibrada.
- 41. En el caso de que el padre sea el portador de una translocación balanceada 21/D y la madre tenga una cariotipo normal, el riesgo de Síndrome de Down por translocación heredada es inferior al 2%. Sin embargo cuando la portadora de la translocación balanceada es la madre, el riesgo de S. de Down en los hijos es del 15%-20%. Cuando la translocación balanceada es entre los cromosomas 21/22 y el portador es el padre el riesgo de hijos downianos es inferior al 2%, mientras que si esta translocación balanceada la presenta la madre el riesgo es del 33%. En el caso de translocación entre homólogos, es decir en la translocación 21/21, tanto si se da en la madre como en el padre, todos los hijos serán downianos por translocación heredada.
- 42. • Cuando encontramos un S. de Down por translocación heredada, debemos examinar a todos los hermanos del paciente, así como a los hermanos del progenitor portador de la translocación balanceada, que aunque fenotípicamente normales pueden ser portadores de la translocación balanceada y por tanto con riesgo elevado de engendrar hijos trisómicos. • Dado que el fenotipo, es decir las manifestaciones clínicas del S. de Down, es idéntico en la trisomía 21 regular y en las translocaciones heredadas y "de novo", es necesario practicar un cariotipo a todo niño/a, afecto de S. de Down.
- 43. Los varones con síndrome de Down se consideran estériles, pero las mujeres conservan con frecuencia su capacidad reproductiva. En su caso también se incrementa la probabilidad de engendrar hijos con SD hasta un 50%, aunque pueden tener hijos sin trisomía.
- 44. Gracias.