Impurities in drug substance (ich q3 a)

This document discusses guidelines for classifying, reporting, controlling, and qualifying impurities in new drug substances. It defines types of impurities and provides thresholds for reporting, identifying, and qualifying impurities. The key points are: - Impurities are classified as organic, inorganic, or residual solvents. Organic impurities can arise from starting materials, byproducts, or degradation. - Identification thresholds determine which impurities must be identified and qualified. Impurities above reporting thresholds must be reported. - Specifications list individual specified impurities and general limits for unspecified impurities. - Qualification involves evaluating safety data for impurities at specified levels. Impurities may need further study if usual qualification thresholds




























Impurities in drug substance (ich q3 a)
- 1. IMPURITIES IN NEW DRUG SUBSTANCES Q3A(R2)
- 2. INTRODUCTION Impurities in new drug substances are addressed from two perspectives: Chemistry Aspects include classification and identification of impurities, report generation, listing of impurities in specifications, and a brief discussion of analytical procedures; and Safety Aspects include specific guidance for qualifying those impurities that were not present, or were present at substantially lower levels, in batches of a new drug substance used in safety and clinical studies.
- 3. DEFINITIONS Impurity: Undesirable element or substance that lowers the quality. Identified Impurity: An impurity for which a structural characterisation has been achieved. Unidentified Impurity: An impurity for which a structural characterisation has not been achieved and that is defined solely by qualitative analytical properties (e.g., chromatographic retention time).
- 4. DEFINITIONS Specified Impurity: An impurity that is individually listed and limited with a specific acceptance criterion in the drug substance specification. A specified impurity can be either identified or unidentified. Unspecified impurity: An impurity that is limited by a general acceptance criterion, but not individually listed with its own specific acceptance criterion, in the drug substance specification. Potential Impurity: An impurity that theoretically can arise during manufacture or storage.
- 5. HIGHLIGHTS CLASSIFICATION OF IMPURITIES REPORTING AND CONTROL OF IMPURITIES ANALYTICAL PROCEDURES REPORTING IMPURITY CONTENT OF BATCHES LISTING OF IMPURITIES IN SPECIFICATIONS QUALIFICATION OF IMPURITIES
- 6. CLASSIFICATION OF IMPURITIES Impurities can be classified into the following categories: Organic impurities (process- and drug-related) Inorganic impurities Residual solvents
- 7. CLASSIFICATION OF IMPURITIES Organic impurities (process- and drug-related) Organic impurities can arise during the manufacturing process and/or storage of the drug substance. Starting materials By-products Intermediates Degradation products Reagents, ligands and catalysts
- 8. CLASSIFICATION OF IMPURITIES Inorganic impurities Inorganic impurities can result from the manufacturing process. Reagents, ligands and catalysts Heavy metals or other residual metals Inorganic salts Other materials (e.g., filter aids, charcoal)
- 9. CLASSIFICATION OF IMPURITIES Residual solvents Solvents are inorganic or organic liquids used as vehicles for the preparation of solutions or suspensions in the synthesis of a new drug substance.
- 10. REPORTING AND CONTROL OF IMPURITIES Organic Impurities Identify the possible & potential impurities most likely to arise during the synthesis, purification, and storage. Stress testing (ICH-Q1A on Stability) used to identify potential impurities arising during storage. Any impurity at a level greater than (>) the identification threshold should be identified. Any degradation impurity observed in stability studies at a level greater than (>) the identification threshold should be identified.
- 11. REPORTING AND CONTROL OF IMPURITIES THRESHOLDS Maximum Daily Dose Reporting Threshold Identification Threshold Qualification Threshold 2g/day 0.05% 0.10% or 1.0 mg per day intake (whichever is lower) 0.15% or 1.0 mg per day intake (whichever is lower) > 2g/day 0.03% 0.05% 0.05% Reporting Threshold : A limit above (>) which an impurity should be reported. Identification Threshold : A limit above (>) which an impurity should be identified. Qualification Threshold : A limit above (>) which an impurity should be qualified.
- 12. REPORTING AND CONTROL OF IMPURITIES Inorganic Impurities Inorganic impurities are normally detected and quantified using pharmacopoeia or other appropriate procedures such as sulphated ash/Residue on ignition. No heavy metals test is recommended due to no longer available from 1st January 2018. Metal(s) used/carryover should be identified and reported as per pharmacopoeia or ICH-Q3D.
- 13. REPORTING AND CONTROL OF IMPURITIES Elemental Impurities Metals are know to be toxic and do not provide any therapeutic benefit so these should be monitored at specified levels. Potential Sources of Elemental Impurities Metal catalysts and metal reagents are used Metals being used in reagents, water, starting materials or excipients through interactions with processing equipment suspected of being leached from container closure system
- 14. REPORTING AND CONTROL OF IMPURITIES Fishbone : Potential Sources of Elemental Impurities
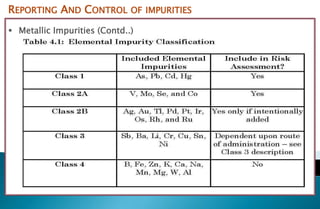
- 15. REPORTING AND CONTROL OF IMPURITIES Metallic Impurities (Contd..) Based on human health risk ICH classified the elemental impurities as Class-1 •Significantly toxic Class-2 •Toxic to a greater or lesser extent based on route of administration Class-3 •Relatively low toxicity (high Permissible Daily Exposure) Class-4 •PDE has not been established due to their low inherent toxicity and/or regional regulations
- 16. REPORTING AND CONTROL OF IMPURITIES Metallic Impurities (Contd..)
- 17. REPORTING AND CONTROL OF IMPURITIES Residual Solvents The control of residues of the solvents used in the manufacturing process for the new drug substance should be discussed and presented according to the ICH Q3C Guideline for Residual Solvents.
- 18. ANALYTICAL PROCEDURES Analytical procedures shall be validated and suitable for the detection and quantification of impurities (ICH Q2A Analytical Validation). Reference standards used in the analytical procedures for control of impurities should be evaluated and characterised for intended use. The drug substance can be used as a standard to estimate the levels of impurities. The quantitation limit for the analytical procedure should be not more than () the reporting threshold.
- 19. REPORTING IMPURITY CONTENT OF BATCHES Quantitative results should be presented numerically, and not in general terms such as “complies”, “meets limit” etc. Any impurity at a level greater than (>) the reporting threshold should be reported. Results should be rounded using conventional rules. Impurities should be designated by code number or by an appropriate descriptor, e.g., retention time. All impurities at a level greater than (>) the reporting threshold should be summed and reported as total impurities.
- 20. LISTING OF IMPURITIES IN SPECIFICATIONS Drug substance specification should include (where applicable): Organic Impurities Each specified identified impurity Each specified unidentified impurity Any unspecified impurity Total impurities Inorganic impurities Residual solvents
- 21. LISTING OF IMPURITIES IN SPECIFICATIONS Individual impurities with specific acceptance criteria should be included in the specification are referred to as "specified impurities“. Note that specified impurities can be identified or unidentified. Specified identified impurities should be included along with specified unidentified impurities estimated to be present at a level greater than (>) the identification threshold. Specified, unidentified impurities should be referred to by an appropriate descriptive label e.g., “unidentified A", “unidentified with RRT of 0.9”. Unspecified impurity should be included with the acceptance criterion of not more than () the identification threshold.
- 22. QUALIFICATION OF IMPURITIES Qualification is the process of acquiring and evaluating data that establishes the biological safety of an individual impurity or a given impurity profile at the level(s) specified. The level of any impurity present in drug substance that has been adequately tested in safety and/or clinical studies would be considered qualified. Impurities that are also significant metabolites present in animal and/or human studies are generally considered qualified.
- 23. DECISION TREE FOR IDENTIFICATION & QUALIFICATION OF IMPURITIES
- 24. NOTES This procedure could not applied for the drug substances: Biological/biotechnological Peptide Oligonucleotide (genetics) Radiopharmaceutical Fermentation product and semi- synthetic products derived therefrom, herbal products Crude products of animal or plant origin
- 25. ? How to fix a limit for specified impurities? Limit can be fixed based on ICH Q6A recommended decision tree # 1.
- 26. A Limit calculated on thresholds can also adopted. Maximum Daily Dose Reporting Threshold Identification Threshold Qualification Threshold 2g/day 0.05% 0.10% or 1.0 mg per day intake (whichever is lower) 0.15% or 1.0 mg per day intake (whichever is lower) > 2g/day 0.03% 0.05% 0.05%
- 27. ? What is skip testing and which parameters can be assumed for skip testing? Definition: Skip testing is the performance of specified tests at predetermined intervals, rather than on a batch-to-batch basis with the understanding that those batches not being tested still must meet all acceptance criteria established. Selection of test parameters: This concept may be applicable to, for example, residual solvents, metallic impurities and microbiological testing (Q6A & Q3D).
- 28. ? Why the Identification & Qualification threshold limit is same for the MDD >2g ? Maximum Daily Dose Reporting Threshold Identification Threshold Qualification Threshold > 2g/day 0.03% 0.05% 0.05% The impurity at Identification threshold should be qualified, due to total daily intake of impurity shall high due to higher dosage, hence the impurities will be identified and qualified at the level of 0.05%.
- 29. ? What will be the limit for an impurity unknown & unidentified, if surviving more than the identification threshold? A level of a qualified impurity higher than that present in a new drug substance can also be justified based on an analysis of the actual amount of impurity administered in relevant safety studies. If data are unavailable to qualify the proposed acceptance criterion of an impurity, studies to obtain such data can be appropriate when the usual qualification thresholds are exceeded.