


















![XVIII
LISTADO DE TABLAS
Tabla 1. Concentraciones típicas normales (µg/L) de agua natural, agua de río y agua
marina ................................................................................................................................. 9
Tabla 2. Normativa vigente en materia de aguas ............................................................... 11
Tabla 3. Concentraciones máximas legisladas para los parámetros y tipos de agua
estudiados ........................................................................................................................... 14
Tabla 4. Métodos de referencia establecidos por la legislación y métodos más utilizados
recientemente ...................................................................................................................... 14
Tabla 5. Hechos históricos referidos a los fenómenos luminiscentes ................................ 18
Tabla 6. Principales reacciones quimioluminiscentes en fase gaseosa .............................. 25
Tabla 7. Principales reacciones quimioluminiscentes en fase líquida ............................... 25
Tabla 8. Principales características de algunos peroxioxalatos ......................................... 32
Tabla 9. Listado de reactivos, casas comerciales y pictogramas de seguridad .................. 34
Tabla 10. Condiciones de derivatización de aminas alifáticas .......................................... 50
Tabla 11. Parámetros de reacción estudiados .................................................................... 53
Tabla 12. Comparación entre la derivatización en cartucho y en disolución .................... 54
Tabla 13. Porcentajes de Recuperación en función del Volumen de muestra procesado .. 54
Tabla 14. Características analíticas para los derivados amina-dansilo con detección UV
o fluorescente ...................................................................................................................... 57
Tabla 15. Estudios de precisión. Repetibilidad (Desviación Estándar Relativa %) .......... 57
Tabla 16. Porcentajes de recuperación y desviación estándar relativa para cada analito
en las diferentes muestras de agua ...................................................................................... 59
Tabla 17. Concentraciones deducidas en la muestra de agua residual (% Error relativo) . 60
Tabla 18. Condiciones optimizadas e intervalos estudiados .............................................. 62
Tabla 19. Condiciones cromatográficas experimentales para la retención,
derivatización, separación y detección en línea de aminas alifáticas .................................
63
Tabla 20. Curvas de calibrado, parámetros analíticos y tiempos de retención para la
derivatización en línea de aminas alifáticas con Dns-Cl .................................................... 64
Tabla 21. Condiciones pre y postcolumna óptimas para la determinación de aminas....... 69
Tabla 22. Parámetros analíticos de los derivados amina-dansilo detectados por
quimioluminiscencia ........................................................................................................... 72
Tabla 23. Limites de detección (µg/l) obtenidos con detección quimioluminiscente a
diferentes volúmenes de muestra y diferentes volúmenes de elución. Comparación con
los valores obtenidos con otros sistemas de detección y reactivos ..................................... 72
Tabla 24. Estudios de precisión (DER %). Repetibilidad y reproducibilidad del método
utilizando diferentes volúmenes de muestra ....................................................................... 73
Tabla 25. Concentración de amina fortificada y concentración de amina fortificada
encontrada (µg/L) en muestras de agua reales .................................................................... 74
Tabla 26. Resumen de algunos procedimientos descritos en la bibliografía para la
determinación de amonio .................................................................................................... 78
Tabla 27. Diseño factorial de optimización de las concentración de reactivos y pH ........ 80
Tabla 28. Importancia e interacción entre los factores: pH, [OPA] y [NAC] ................... 81
Tabla 29. Curvas de calibrado de amonio y parámetros analíticos utilizando el reactivo
OPA-NAC .......................................................................................................................... 81
Tabla 30. Curvas de calibrado de amonio y parámetros analíticos utilizando el Método
de Nessler ........................................................................................................................... 82](https://image.slidesharecdn.com/meseguer-170216182421/85/Meseguer-20-320.jpg)
![XIX
Tabla 31. Concentración de amonio hallada en muestras reales utilizando los diferentes
métodos estudiados ............................................................................................................. 86
Tabla 32. Curvas de adición estándar para muestras reales utilizando el Método OPA-
NAC .................................................................................................................................... 87
Tabla 33. Propiedades analíticas de los tres métodos estudiados ...................................... 88
Tabla 34. Comparación del procedimiento de derivatización en disolución establecido
por Marcé y col. [67] y el procedimiento de derivatización en disolución optimizado ..... 92
Tabla 35. Ecuaciones de las curvas de calibrado (Atura de pico (Voltios) frente a
Concentración de NH4
+
(mg/L)) obtenidas en diferentes condiciones ............................... 93
Tabla 36. Estudios de precisión (%DER) y exactitud (%Er) proporcionados por el
método a diferentes niveles de concentración de amonio (n, número de réplicas) ............ 94
Tabla 37. Test t de comparación de pendientes de las ecuaciones del MOSA para todas
las muestras evaluadas con la pendiente de la curva de calibrado de patrones de amonio.
Los valores tabulados corresponden a un nivel de confianza del 95 % excepto *(98 %) y
**(99 %) ............................................................................................................................. 96
Tabla 38. Concentración de amonio encontrada en muestras reales fortificadas con
amonio a diferentes niveles de concentración antes y después del tratamiento Kjeldahl .. 97
Tabla 39. Respuesta de screening y Concentración de amonio deducida para las
muestras estudiadas ............................................................................................................ 98
Tabla 40. Comparación de sensibilidad de los detectores en la reacción QL
Cr(III)/luminol/H2O2 .......................................................................................................... 101
Tabla 41. Optimización de las variables químicas y físicas del sistema de inyección en
flujo ..................................................................................................................................... 102
Tabla 42. Curvas de calibrado de Cu(II) y parámetros analíticos en diferentes
condiciones (IL: Intervalo lineal; LD: Límite de detección) .............................................. 103
Tabla 43. Estudio de la interferencia de iones metálicos ................................................... 105
Tabla 44. Resultados obtenidos en el screening y determinación de Cu(II) en muestras
de agua reales ...................................................................................................................... 106
Tabla 45. Estudios recientes basados en la reacción del luminol-H2O2 para cromo ......... 111
Tabla 46. Curvas de calibrado para Cr(III) y Cr(VI) ......................................................... 112
Tabla 47. Curvas de calibrado en el intervalo lineal para las mezclas de Cr(III)+Cr(VI) . 113
Tabla 48. Parámetros analíticos para las determinaciones de Cr(III), Cr(VI) y Cr Total .. 114
Tabla 49. Curvas de calibrado simuladas y reales utilizando modelos de calibración
polinómicos ........................................................................................................................ 116
Tabla 50. Pendientes y porcentajes de recuperación para la muestra de HCl ................... 116
Tabla 51. Determinación de Cromo en una muestra de agua mineral ............................... 117
Tabla 52. Estudios de la determinación quimioluminiscente en flujo de iones metálicos
en muestras de agua ............................................................................................................ 120
Tabla 53. Composición del material de referencia certificado SRM © 1640: agua dulce. 121
Tabla 54. Parámetros analíticos para la determinación de Cr(III), Co(II) y Cu(II) en
muestras no acidificadas. Procedimiento en flujo .............................................................. 123
Tabla 55. Parámetros analíticos de las curvas de calibrado de patrones no acidificados
utilizando el procedimiento en estático .............................................................................. 124
Tabla 56. Parámetros analíticos para la determinación de Cr(III), Co(II) y Cu(II) en
muestras acidificadas con HCl 5· 10
-3
M. Procedimiento en flujo ...................................... 125
Tabla 57. Parámetros analíticos para la determinación de Cr(III), Co(II) y Cu(II) en
muestras acidificadas con HNO3 0.5 M. Procedimiento en flujo ....................................... 126
Tabla 58. Modelos PLS en el intervalo lineal .................................................................... 133
Tabla 59. Modelos de PLS en el intervalo no lineal .......................................................... 134](https://image.slidesharecdn.com/meseguer-170216182421/85/Meseguer-21-320.jpg)











![Capítulo 1. INTRODUCCIÓN
9
1.1. CONSIDERACIONES SOBRE EL ANÁLISIS DE AGUA Y
ANALITOS ESTUDIADOS
1.1. CARACTERÍSTICAS DE LAS AGUAS
La ciencia del medio ambiente comprende el estudio completo del entorno que
nos rodea y por ello necesita del apoyo de todas las ciencias, siendo por tanto
multidisciplinar. Un gran número de estudios, muestran la necesidad de conocer la
composición de las partes del medio implicado. No es posible por ejemplo, estudiar el
transporte de sustancias en el ciclo del agua, sin medir las sustancias que están siendo
transportadas.
La composición química de las aguas naturales depende entre otras de variables
como el tipo de agua, localización geográfica o estación del año [1]. Los constituyentes
mayoritarios (1-1000 mg/L) son Na+
, Ca2+
, Mg2+
, HCO3
-
, SO4
2-
y Cl-
, y las especies
minoritarias (0.1-10 mg/L) Fe2+
, Fe3+
, Sr2+
, K+
, CO3
2-
, NO3
-
, F-
, H3BO3. La presencia
natural de otras especies ocurre en un nivel de concentraciones inferior, como se expone
en la Tabla 1.
Tabla 1: Concentraciones típicas normales (µg/L) de agua natural, agua de río y agua
marina.
Elemento Natural Río Mar Elemento Natural Río Mar
Li 10 12 200 As 0.5 2 3
B 10 10 5000 Se 0.2 0.2 0.1
Al 10 50 2 Br 15 20 7000
Ti 5 10 1 Rb 1 20 120
V 0.5 1 2.5 Sr 70 60 8000
Cr 1 1 0.05 Mo 0.5 1 10
Mn 10 7 0.2 Cd 0.03 0.02 0.1
Fe 500 40 2 Sb 0.2 0.3 0.2
Co 0.05 0.2 0.02 I 2 10 60
Ni 0.5 0.3 0.05 Cs 0.02 0.04 0.1
Cu 3 5 2 Hg 0.07 0.007 0.03
Zn 15 20 10 Pb 1 3 0.03
Esta composición puede ser alterada por fuentes naturales o antropogénicas. La
contaminación del medio hídrico es un problema grave y con serias consecuencias que
ha afectado a numerosas zonas a pesar de la auto purificación del propio ciclo. El
resultado final es que la composición de las aguas está alterada por la presencia de
compuestos inorgánicos y orgánicos adicionales [1-3]. El ciclo del agua se verá afectado
por el aumento de la demanda biológica de oxígeno y los procesos de eutrofización, y
también por la acidificación y salinización de las aguas [4].](https://image.slidesharecdn.com/meseguer-170216182421/85/Meseguer-33-320.jpg)
![CONSIDERACIONES SOBRE EL ANÁLISIS DE AGUA Y ANALITOS ESTUDIADOS
10
Existen características específicas para cada tipo de agua contaminada:
§ Las aguas residuales urbanas e industriales son disoluciones acuosas complejas que
contienen una variada gama de compuestos orgánicos e inorgánicos, tanto disueltos
como en suspensión y también microorganismos [1]. Pueden contener
concentraciones puntuales muy elevadas de contaminantes, lo que puede originar
variaciones muy significativas en los equilibrios originando especies nuevas.
§ Las aguas subterráneas presentan problemas de salinidad debido a procesos de
infiltración del agua marina, y también de toxicidad debido al almacenamiento de
sustancias tóxicas en tanques subterráneos o a actividades agrícolas.
§ Las aguas costeras reciben la descarga directa de los ríos, vertidos de barcos o
liberación de materiales de protección de embarcaciones. Presentan problemas de
eutrofización, de bioacumulación de metales tóxicos en los moluscos o
contaminación microbiana. Los problemas a niveles profundos se deben a la difusión
desde la superficie o desde los sedimentos por lo que se estudian tanto gradientes
horizontales como verticales, ya que ambos definen su nivel de impacto y su
biodisponibilidad.
Dada la gran variedad de muestras de agua medioambientales y los problemas
que presentan, en esta Tesis se han estudiado diferentes tipos de agua, con el fin de
abarcar una parte de la amplia variabilidad que nos podemos encontrar en cuanto a la
matriz. Se han estudiado aguas: embotelladas, de grifo, de riego, de fuente, de lago,
residuales y marinas.
La legislación vigente en materia de aguas comprende por una parte las
Directivas Europeas, y por otra, las Órdenes, Leyes y Reales Decretos tanto estatales
como autonómicos. En la Tabla 2, se puede ver la normativa más representativa en los
diferentes ámbitos en materia de aguas. Las normativas estatales y autonómicas se ajustan
a las exigencias de la normativa europea, siendo igual o más restrictivas que ésta.](https://image.slidesharecdn.com/meseguer-170216182421/85/Meseguer-34-320.jpg)

![CONSIDERACIONES SOBRE EL ANÁLISIS DE AGUA Y ANALITOS ESTUDIADOS
12
1.2. CARACTERÍSTICAS DE LOS ANALITOS ESTUDIADOS Y
LEGISLACIÓN APLICABLE A LOS MISMOS.
Se han estudiado analitos de diferentes tipos; metales como Cr(III), Cr(VI),
Co(II) y Cu(II), amonio y nitrógeno total, aminas alifáticas primarias y secundarias
(metilamina (MA), etilamina (EA), butilamina (BA), pentilamina (PeA), hexilamina (HA),
dimetilamina (DMA), dietilamina (DEA) y β-feniletilamina (β-FEA)), y poliaminas
(Putrescina (Put), Cadaverina (Cad), Espermina (Spm) y Espermidina (Spd)).
1.1.2.1. Descripción de los analitos estudiados
Los metales se introducen en el ciclo del agua básicamente por residuos
industriales. Muchos de estos iones son tóxicos para el hombre y sus emisiones han de
monitorizarse y controlarse.
El Cromo es un contaminante común en aguas naturales y residuales y lo
podemos encontrar como Cr(III) o Cr(VI). Según el estado de oxidación de un elemento,
su biodisponibilidad y toxicidad varían drásticamente, de hecho, el Cr(VI) es mucho más
tóxico que el Cr(III). El Cr(III) no es tóxico a bajos niveles y se considera esencial en
mamíferos. La toxicidad del Cr(VI) como aerosol es bien conocida: produce daños en la
piel y sistema respiratorio, y puede producir cáncer [5]. Sin embargo, los efectos tóxicos
del Cr(VI) en agua no están bien documentados.
La contaminación con Cr(III) y Cr(VI) resulta de los vertidos de industrias de
curtidos, acero, tintes o de sectores de fabricación de productos como pinturas, pigmentos
o fungicidas. Este metal puede introducirse en aguas potables debido a los inhibidores de
corrosión utilizados en cañerías o recipientes.
El Cobalto se encuentra en las rocas, el suelo, el agua, plantas y animales.
Existen formas radioactivas y no radioactivas de cobalto. El cobalto no radioactivo,
llamado cobalto estable, se usa para producir aleaciones (mezclas de metales) usadas en
la manufactura de motores de aviones, imanes, herramientas para triturar y cortar,
articulaciones artificiales y también para colorear vidrio, cerámicas y pinturas y como
secador de esmaltes y pinturas para porcelana.
La población general está expuesta a bajos niveles de Cobalto en el aire, el agua y
los alimentos. El cobalto tiene efectos tanto beneficiosos como perjudiciales sobre la
salud. En bajos niveles, es parte de la vitamina B12, sustancia que es esencial para
mantener una buena salud. A niveles altos, puede dañar los pulmones y el corazón [6].
El Cobre se emplea en alambres y cables eléctricos y en algunas cañerías de
agua. También forma parte de aleaciones como latón y bronce. Las sales de Cobre se
utilizan comúnmente en la agricultura para tratar enfermedades de las plantas, como el
moho, para tratar agua, y como preservativos para alimentos, cueros y telas.
Bajos niveles de cobre son esenciales para el hombre, mientras que niveles altos
pueden producir efectos nocivos tales como irritación de la nariz, la boca y los ojos,
vómitos, diarrea, calambres estomacales y náuseas [7].](https://image.slidesharecdn.com/meseguer-170216182421/85/Meseguer-36-320.jpg)
![Capítulo 1. INTRODUCCIÓN
13
El amonio es uno de los productos químicos más ampliamente utilizados. Se
utiliza principalmente como fertilizante, y también en la fabricación de fibras, explosivos
o plásticos y como alimento de animales. También se usa en insecticidas, en la
fabricación de detergentes y limpiadores y como refrigerante [8].
Es el principal contaminante que provoca procesos de eutrofización en los
sistemas acuosos, y por eso, su concentración en aguas de consumo humano viene
regulada por la legislación.
Las aminas alifáticas de bajo peso molecular, son intermediarios importantes en
las industrias químicas y farmacéuticas. Algunas de ellas se producen en cantidades de
más de 100000 toneladas por año en el Oeste de Europa. Debido a su carácter polar, son
difíciles de eliminar de los sistemas acuosos. Además de su aplicación industrial, las
podemos encontrar como productos de degradación de materia orgánica como las
proteínas y aminoácidos u otros compuestos de nitrógeno. Las aminas alifáticas
secundarias pueden reaccionar con nitrito produciendo nitrosaminas carcinogénicas.
Hasta ahora, existe muy poca información acerca de la presencia de aminas
alifáticas en aguas residuales industriales y en aguas superficiales, pero se sabe que en
algunos tipos de agua aparecen en cantidades muy pequeñas, por lo que en este caso, los
métodos para su análisis deben ser suficientemente sensibles.
Las aminas biogénicas son bases orgánicas que se forman y degradan como
resultado de la actividad metabólica en plantas, animales y microorganismos. Puede
hallarse en fluidos biológicos, en muestras medioambientales y en corrientes de procesos
industriales normalmente a niveles de trazas [9].
1.1.2.2. Legislación aplicable
Los avances en la determinación de estos u otros analitos, vendrán marcados por
la legislación vigente, que con la evolución continua de los métodos de análisis, tiende a
la reducción progresiva de los valores permitidos y a la exigencia de calidad en los
resultados generados, priorizando la acreditación y validación de los laboratorios.
La concentración de los analitos estudiados en esta Tesis, viene legislada en
función del tipo de agua analizada. Para las aminas, no existe una concentración máxima
permitida establecida por la legislación, pero sí lo está la concentración del Nitrógeno
Kjeldahl Total (TKN). En la Tabla 3, se señalan las concentraciones máximas
permitidas en cada caso. Actualmente, no existe normativa que regule la calidad exigida
a las aguas superficiales destinadas a riego, por lo que se considerará la misma legislación
que para aguas superficiales destinadas a potabilización.
La legislación establece además los métodos de referencia para cada uno de los
parámetros cuya concentración limita. En la Tabla 4, se reflejan los métodos de análisis
establecidos para los analitos en estudio así como los métodos utilizados recientemente en
Química Analítica en muestras de agua.](https://image.slidesharecdn.com/meseguer-170216182421/85/Meseguer-37-320.jpg)

![Capítulo 1. INTRODUCCIÓN
15
1.2. CARACTERÍSTICAS Y TENDENCIAS DEL ANÁLISIS
MEDIOAMBIENTAL
Los avances tecnológicos y metodológicos han conducido a una evolución de la
Química Analítica en general y del campo del análisis del agua en particular. Este hecho
se refleja en el porcentaje de publicaciones científicas (aproximadamente un 54%) que se
ha dedicado a las determinaciones en matrices acuosas dentro del campo medioambiental
(revisión bibliográfica en Analytical Abstract en el período 1980-2001 [10]).
Los propósitos generales del análisis medioambiental [4]son la monitorización
del fondo (definido como el intervalo de concentraciones en el agua de un cierto
elemento, especie o sustancia derivado de fuentes biológicas, geológicas o atmosféricas
naturales) y la monitorización de la contaminación (determinar la concentración de
constituyentes dañinos de origen antropogénico).
Los problemas que surgen en el análisis de aguas son: las bajas concentraciones
de los analitos, la complejidad de la matriz, la presencia de interferentes, la variabilidad
de las muestras o la especiación entre otros. En la actualidad, nos encontramos en un
período de evolución de la QA medioambiental, caracterizado por el desarrollo de nuevas
y mejores técnicas analíticas. El protocolo del proceso analítico dependerá del problema a
analizar y requerirá un estudio de la sensibilidad, exactitud, precisión, fiabilidad,
interferencias, efectos matriz, costes y tiempo de análisis, etc. teniendo siempre en cuenta
factores como la validación, representatividad, impacto medioambiental y toxicidad de
los reactivos.
El estudio de analitos a niveles de trazas, requiere un mayor esfuerzo para
garantizar la calidad del análisis. Cuanto mayores sean la precisión y exactitud exigidas,
más estricto deberá ser el control de calidad. En este sentido, se han elegido los métodos
quimioluminiscentes para la realización de esta Tesis puesto que proporcionan una
elevada sensibilidad en un amplio intervalo dinámico de concentraciones, con una
instrumentación simple (no requieren fuente de excitación), amplia versatilidad y se
acoplan fácilmente como métodos de detección a sistemas de análisis por inyección en
flujo o sistemas de separación cromatográficos.
Como perspectiva de futuro, se han establecido una serie de analitos prioritarios
para los que se deben desarrollar estudios ambientales, proponer métodos alternativos o
mejorar los existentes [11]. Entre ellos, se encuentran los metales pesados, el amonio y el
nitrógeno total, parámetros que han sido objeto de estudio en esta Tesis.
Los avances metodológicos y químicos, vendrán marcados por la búsqueda de
límites de detección cada vez más bajos y el empleo de sensores o el desarrollo de
técnicas sencillas, de rápida respuesta, bajo coste y sin necesidad de personal
especializado para la vigilancia. La tendencia más marcada es a la completa
automatización, destacando la utilización de sistemas de inyección en flujo, así como la
automatización de sistemas en HPLC. La derivatización de 7 aminas alifáticas con
Cloruro de Dansilo en cartuchos C18 y posterior separación cromatográfica con](https://image.slidesharecdn.com/meseguer-170216182421/85/Meseguer-39-320.jpg)
![CARACTERÍSTICAS Y TENDENCIAS DEL ANÁLISIS MEDIOAMBIENTAL
16
detección fluorescente (Apéndice 1), o quimioluminiscente por reacción postcolumna de
los derivados dansilados con TCPO/H2O2 (Apéndice 3), se han desarrollado en esta
Tesis. Para la detección fluorescente, se ha logrado la automatización completa del
análisis, consiguiendo la derivatización en línea acoplada a la separación
cromatográfica (Apéndice 2).
Se ha propuesto un nuevo método fluorescente para la determinación de amonio
en muestras de agua, basado en la reacción del amonio con el reactivo OPA-NAC,
comparándose los resultados con los obtenidos con el método de referencia de Nessler y
el método del electrodo selectivo que han sido revisados previamente a su utilización
(Apéndice 4). Como complemento, y con la intención de mejorar la sensibilidad, se
propone un método quimioluminiscente para la determinación de amonio derivatizando
este en disolución con Cloruro de Dansilo y separándolo de posibles interferentes
mediante HPLC y posterior generación de quimioluminiscencia con TCPO/H2O2
(Apéndice 5). Un estudio similar se ha realizado para la determinación del Nitrógeno de
grupos amino primario más Nitrógeno amoniacal, mediante la aplicación del método de
fluorescencia por derivatización con el reactivo OPA-NAC (Apéndice 10), o por
detección quimioluminiscente basada en la reacción del Nitrógeno Orgánico tipo amino
y amonio con Hipoclorito, y medida de la disminución de señal quimioluminiscente al
reaccionar con el luminol (Apéndice 11).
Además, se ha extendido el uso de métodos de screening [12-13], que permiten
seleccionar las muestras que superan el valor legislado, para posteriormente analizarlas
por el método propuesto si está validado, o por el método de referencia que esté
establecido. En esta dirección, se ha propuesto un método de análisis por inyección en
flujo para el screening de muestras de agua para Cu(II), basado en la disminución de la
señal quimioluminiscente emitida por el sistema Coproporfirina I/TCPO/H2O2 en
presencia de Cu(II) (Apéndice 6).
La especiación de elementos metálicos presenta gran relevancia debido a que las
diferentes especies presentan también diferentes características en cuanto a toxicidad,
biodisponibilidad o reactividad. En esta Tesis, se estudia la exactitud del método de
análisis por inyección en flujo para la especiación de Cr(III) y Cr(VI) basado en la
reacción quimioluminiscente del luminol con H2O2 (Apéndice 7). Como complemento a
este trabajo se ha abordado también la influencia del protocolo de acondicionamiento de
las muestras de agua en la detección quimioluminiscente de elementos traza (Apéndice
8). Además, basándonos en esta misma reacción, se propone un método de calibración
multivariante para la determinación simultánea de Cr(III) y Co(II) en estático
(Apéndice 9).
Los parámetros básicos que definen un resultado analítico son la exactitud o
proximidad al valor verdadero, y la incertidumbre expresada como el intervalo de
confianza. Para asegurar la calidad de los resultados medioambientales existen
diferentes metodologías:
§ Empleo de buenas prácticas de laboratorio: Consiste en realizar las distintas
operaciones del laboratorio en las mejores condiciones de calidad y seguridad.](https://image.slidesharecdn.com/meseguer-170216182421/85/Meseguer-40-320.jpg)

![QUIMIOLUMINISCENCIA
18
1.3. QUIMIOLUMINISCENCIA
1.3.1. PERSPECTIVA HISTÓRICA
La observación e investigación de los fenómenos luminiscentes tiene una larga
historia. Desde tiempos inmemoriales, se conocían sustancias y animales que
resplandecían en las sombras, por lo que despertaban la curiosidad y las supersticiones
[14]. En la Tabla 5, se describen los principales hechos históricos que han propiciado la
investigación de este tipo de fenómenos.
Tabla 5. Hechos históricos referidos a los fenómenos luminiscentes.
Año Investigador Hecho
1500 -1000
a.C.
Crónicas chinas
Shih Ching
Primeras referencias escritas sobre luciérnagas y gusanos
luminiscentes (Libro de las Odas).
384 - 322
a.C.
Aristóteles
Observó que la luz emitida se producía sin calentamiento.
23 – 79
d.C.
Caius Plinius
Secundus
Describió con detalle un gran número de organismos
luminosos.
1555 Conrad Gesner
Publicó un libro cuyo contenido trataba exclusivamente de
los fenómenos luminiscentes.
1565 Nicolás Monarde
Escribió acerca del extraordinario color azul intenso de un
extracto acuoso de la madera llamada “lignum
nephrilicum”.
1603 Vincenzo Cascariolo
Introdujo la luminiscencia de los sólidos. Calentó polvos
de sulfato de bario con carbón y encontró que la mezcla
resultante brillaba en la noche. La piedra se “cargaba” de
luz solar por el día y brillaba durante horas en la oscuridad
(fosforescencia). Le llamó lapis solaris (piedra del Sol).
1652 Nicolas Zucchi
Demostró, por medio de filtros ópticos, que el color de la
luz emitida durante la noche era la misma que cuando la
piedra era expuesta a luz blanca o de otros colores, como
azul o verde.
1655
Athanasius Kircher,
Francisco Grimaldi,
Isaac Newton y
Robert Boyle
Observaron que cuando el extracto acuoso de la madera
“lignum nephrilicum” era ilu minada con luz blanca,
aparecía una luz reflejada azul intensa, mientras que la luz
transmitida era amarilla.
1668 Robert Boyle
Marcó inicialmente las diferencias esenciales entre
incandescencia y luminiscencia (emisión de luz por
materiales calentados o no calentados respectivamente).
1845 George Stokes
Caracterizó la naturaleza bicromática del cristal de fluoruro
cálcico como una emisión (fosforescencia). Demostró que
la luz incidente de una región espectral era absorbida y
transformada por la solución en luz emitida en una región
espectral diferente, de mayor ë (Ley de Stokes). La
emisión desaparecía instantáneamente cuando se apagaba
la luz incidente. Le dio el nombre de fluorescencia.](https://image.slidesharecdn.com/meseguer-170216182421/85/Meseguer-42-320.jpg)
![Capítulo 1. INTRODUCCIÓN
19
1877 Radziszewski
Observó por primera vez la emisión de luz causada por
reacciones químicas [15] (QUIMIOLUMINISCENCIA)
al burbujear con O2 una disolución etanólica alcalina de
lofina.
1888 Eilhard Wiedemann
Introdujo el término luminiscencia para describir la
emisión de luz que no requería un aumento de la
temperatura, abarcando la fluorescencia y la
fosforescencia.
1927 Albrecht
Describió las propiedades quimioluminiscentes del
luminol[16].
1935 Gleu y Petsch
Describieron la luz azul o verde emitida por el nitrato
bis(N-Metilacridinio) (lucigenina) [17].
1964 McCapra
Propuso un mecanismo basado en la formación de un ciclo
de dioxoetanona para explicar la quimioluminiscencia de
las sales acridínicas [18].
Hoy en día, la luminiscencia se entiende como el proceso por el cual un material
genera radiación no térmica (depende de las características del tipo de material) [19]. Así,
la luminiscencia es la emisión de luz por medios diferentes a la combustión y por eso
ocurre a temperaturas más bajas que las requeridas por la combustión. La intensidad de
emisión luminiscente es baja si se compara con las fuentes incandescentes, pero tiene la
ventaja de que habitualmente emana de cantidades de materia extremadamente pequeñas.
Esto proporciona implicaciones beneficiosas para las ciencias analíticas, sin embargo, el
uso de la luminiscencia para el análisis es bastante reciente.
Registrando la intensidad de luminiscencia relativa en función de la
concentración, se pueden determinar cuantitativamente (frecuentemente a niveles de
trazas) un amplio rango de analitos tanto inorgánicos como orgánicos. La luminiscencia,
generalmente ofrece superior selectividad, detectabilidad y rango lineal comparado con la
espectrometría de absorción.
Muchas publicaciones analíticas muestran que la metodología fluorimétrica es
más común que la fosforescencia o la quimioluminiscencia. Sin embargo, los trabajos de
investigación indican un aumento en la aceptación de estas dos últimas técnicas
espectroscópicas, específicamente cuando se combinan con separaciones cromatográficas
[20-24].
Las investigaciones sobre el potencial analítico de la quimioluminiscencia (QL)
para análisis de rutina datan de los años 70 para las reacciones en fase gaseosa, y de la
década de los 80 para las reacciones en fase líquida. Hoy en día el interés analítico en la
aplicación de la QL está creciendo exponencialmente.](https://image.slidesharecdn.com/meseguer-170216182421/85/Meseguer-43-320.jpg)



![Capítulo 1. INTRODUCCIÓN
23
1.3.3.2. Tendencia Actual
La importancia de las determinaciones quimioluminiscentes en el campo de la
Química Analítica, ha tenido una evolución creciente en los últimos años. Durante los
años 70 y 80, la investigación en este tipo de reacciones estaba orientada a la búsqueda de
nuevos reactivos quimioluminiscentes [25-27] y el estudio de reactivos estructuralmente
semejantes a los que ya se conocían como quimioluminiscentes.
A partir de los años 90 y hasta la actualidad, los estudios de reacciones
quimioluminiscentes se han dirigido más hacia la mejora en la instrumentación,
construyendo detectores muy sencillos para este tipo de medidas [28-30], y también a la
obtención de estos sistemas de detección acoplados a técnicas separativas permitiendo la
resolución y cuantificación de varios analitos en mezclas complejas por ejemplo de
ácidos[31], compuestos de nitrógeno [32], o biopolímeros [33]. Las grandes aplicaciones
de la QL como método de detección en flujo, cromatografía líquida o electroforesis
capilar, junto con el gran potencial del inmunoensayo, hacen de la técnica un campo de
investigación muy interesante en una amplia variedad de disciplinas.
La detección quimioluminiscente es una técnica en pleno desarrollo. El interés de
la QL en Química Analítica, queda reflejado en el número de publicaciones científicas en
revistas de gran prestigio (Analytical Chemistry, Analytica Chimica Acta, The Analyst,
Talanta, Journal of Chromatography y Journal of pharmaceutical and Biomedical
Analysis entre otras) que, según la base de datos Analytical Abstracts desde 1979 hasta la
actualidad (finales de 2003), es del orden de 3700 publicaciones. La Figura 3 refleja el
crecimiento exponencial en el número de publicaciones durante estos últimos 25 años.
Figura 3: Número de publicaciones en revistas científicas de ámbito internacional frente a
cada año desde 1979 a 2003.
0
100
200
300
1979 1982 1985 1988 1991 1994 1997 2000 2003
Año
NºPublicaciones](https://image.slidesharecdn.com/meseguer-170216182421/85/Meseguer-47-320.jpg)
![QUIMIOLUMINISCENCIA
24
1.3.4. REACCIONES QUIMIOLUMINISCENTES
La bibliografía muestra la aplicación de reacciones quimioluminiscentes en fase
sólida, líquida y gaseosa.
Las reacciones en fase sólida han sido las menos aplicadas pues son reacciones
muy limitadas por la débil emisión que acompaña a la oxidación de muchos materiales
orgánicos. Los sólidos se calientan en presencia de oxígeno y la emisión QL es
proporcional a la velocidad de reacción. En ausencia de oxígeno, la emisión QL se debe a
la descomposición de los peróxidos formados. Se puede recoger la emisión directa de
estas especies o la indirecta (exaltando la QL por adición de fluoróforos). En este campo,
se han descrito reactores para la detección quimioluminiscente en fase sólida, utilizando
como soportes metacrilatos polimerizados. Un ejemplo es la reacción de perioxalato con
peróxido de hidrógeno que utiliza como fluoróforo el 3-aminofluoranteno inmovilizado
en el soporte [34].
Los métodos quimioluminiscentes en fase gaseosa, se han utilizado combinados
con cromatografía gaseosa (utilizando por ejemplo la reacción quimioluminiscente del
ozono [35]), cromatografía de fluidos supercríticos (reacciones del nitrógeno y el sulfuro
[36-38]), y también con detectores de gases no cromatográficos en los que se requiere una
respuesta rápida como el detector quimioluminiscente de monitorización continua de
sulfuro [39]. Algunos de los analitos que se han determinado mediante esta técnica son:
óxidos de nitrógeno (NOx), ozono, y peróxido de hidrógeno en muestras atmosféricas, y
detección y secuenciación de DNA, ácidos nucleicos, fosfatasa alcalina yoxidantes en
riñones, cerebro y plasma. En la Tabla 6, se pueden observar las principales reacciones
en fase gas [40] y las condiciones de reacción necesarias en cada caso, siendo las más
aplicadas las utilizadas para la determinación de azufre, fósforo y nitrógeno total. En
algunos casos la reacción se da a temperatura ambiente, y en otros, se requiere
calefacción a elevadas temperaturas.
Las reacciones quimioluminiscentes en fase líquida, tienen aplicabilidad en la
determinación de un amplio rango de analitos, desde elementos metálicos como cromo y
cobalto a niveles traza [41-42], hasta fármacos como tiopronina [43] o azitromicina [44].
En la Tabla 7, se reflejan las principales reacciones quimioluminiscentes utilizadas en
fase líquida para diferentes analitos estudiados [40,45]. Se dividirán estas reacciones
según la QL sea directa o indirecta. Dado que las reacciones quimioluminiscentes
estudiadas en esta Tesis, son en fase líquida, se detallará más ampliamente este apartado.](https://image.slidesharecdn.com/meseguer-170216182421/85/Meseguer-48-320.jpg)

![QUIMIOLUMINISCENCIA
26
1.3.5. REACCIONES QUIMIOLUMINISCENTES DIRECTAS EN FASE
LÍQUIDA
1.3.5.1. Reacciones del Luminol y sus análogos
El luminol (5-amino-2,3-dihidro-1,4-ftalazindiona), es uno de los reactivos
quimioluminiscentes más comúnmente empleado en fase líquida. Fue presentado por
primera vez en 1928, y la caracterización de la cinética de reacción y las especies
emisoras requirió más de tres décadas de investigación. El luminol se oxida casi
cuantitativamente a 3-aminoftalato en medio básico tanto en disolventes polares como
apolares. El mecanismo propuesto para la reacción [45] es el que se muestra en la Figura
4.
Figura 4. Mecanismo propuesto para la reacción del luminol.
Como oxidantes de la reacción, se han utilizado: el permanganato en la
determinación de paracetamol en fármacos [46] o de urea en tejidos de plantas [47]; el
hipoclorito, en la determinación de amonio [48] o catecolaminas [49]; y el yodo, en la
determinación de penicilinas [50-51]; pero el oxidante más utilizado ha sido el peróxido
de hidrógeno.
O2
O
ONH2
NH
NH OH-
O2
O
ONH2
N
N
O
ONH2
N
N
O
O
-
-
+ N2
O
ONH2
O
ONH2
O
O
-
-
*
O
ONH2
O
ONH2
O
O
-
- + hν (λ = 425 nm)
Luminol
3-aminoftalato](https://image.slidesharecdn.com/meseguer-170216182421/85/Meseguer-50-320.jpg)
![Capítulo 1. INTRODUCCIÓN
27
Cuando la reacción del luminol con peróxido de hidrógeno se da en medio
orgánico (en dimetilsulfóxido), no se requiere ningún catalizador, pero cuando se da en
medio acuoso, se requiere la presencia de un catalizador. Como catalizadores, se han
utilizado diferentes metales de transición (Co2+
, Cu2+
, Fe3+
, Cr3+
,...) [52-54], ferricianuro
[55-56], o algunos metalocomplejos (hemina, hemoglobina y peroxidasas [57-58]). La
intensidad de emisión quimioluminiscente tiene un máximo a 425 nm cuando se trabaja
en medio acuoso, y a 500 nm cuando se trabaja en dimetilsulfóxido. El pH óptimo de la
reacción está entre 8 y 11, dependiendo del catalizador.
La reacción se ha utilizado para la determinación de los catalizadores (límites de
detección desde 1 µM hasta 1 nM según las especies), del H2O2 (límite de detección 0.1
µM) o del luminol (límite de detección 1 pM) [59-60].
También se puede iniciar la reacción QL electroquímicamente, sobre un electrodo
de Ag/AgCl a +0.5V [61-62]. Los límites de detección con QL electrogenerada son
similares a los obtenidos con un catalizador convencional.
En los últimos años, se han revisado compuestos de estructura similar al luminol
con la intención de mejorar el rendimiento cuántico, y se ha concluido que la menor
alteración en el anillo heterocíclico destruye las propiedades quimioluminiscentes de la
molécula.
El luminol se puede enlazar a algunos analitos para inmunoensayo mediante la
sustitución de los hidrógenos del grupo amino primario; esta sustitución provoca una
disminución en la intensidad de emisión quimioluminiscente.
Compuestos derivados del luminol como el isoluminol (4-aminoftalhidrazida)
[63-64] también presentan propiedades QL pero son menos eficaces. Otro derivado del
luminol, el ABEI (aminobutiletilisoluminol) [65-66] se utiliza como reactivo marcador.
En esta Tesis, se presentará una guía para evitar los errores sistemáticos en la
determinación quimioluminiscente de Cr(III) y Cr(VI) en medio acuoso, utilizando la
reacción del luminol con H2O2 y un sistema de análisis por inyección en flujo (Apéndice
7). Se revisará también la influencia de los protocolos de acondicionamiento de la
muestra en el análisis quimioluminiscente de elementos metálicos a niveles de trazas
(Apéndice 8). Por otra parte, se aplicará esta misma reacción en estático, proponiendo
un modelo de calibración multivariada para la determinación quimioluminiscente
simultánea de Cr(III) y Co(II) (Apéndice 9). En todos los casos, se abordará el análisis
de muestras de agua.
También se propone la utilización de la reacción del luminol con Hipoclorito
sódico como oxidante en medio básico. Se determinará la cantidad de Nitrógeno
Orgánico (aminas y aminoácidos) y Amoniacal en muestras de agua basándose en la
disminución de la señal QL por reacción de los compuestos de nitrógeno con hipoclorito
para dar cloraminas (Apéndice 11). La determinación QL se comparará con la
estimación del Nitrógeno Kjeldahl Total por digestión-destilación y detección](https://image.slidesharecdn.com/meseguer-170216182421/85/Meseguer-51-320.jpg)
![QUIMIOLUMINISCENCIA
28
fluorescente basada en la reacción del amonio con el reactivo OPA/NAC o UV/vis
basada en la reacción del amonio con el reactivo de referencia de Nessler (Apéndice 10).
1.3.5.2. Reacción de los ésteres de acridinio
Los ésteres de acridinio reaccionan con peróxido de hidrógeno en medio alcalino
dando lugar a N-Metilacridona en estado excitado que emite luz a 440 nm. El mecanismo
propuesto para esta reacción [40] se muestra en la Figura 5, no se requiere de
catalizadores. Se ha utilizado para la determinación cuantitativa de peróxido de hidrógeno
[67], pero la principal aplicación de estos ésteres radica en su uso como marcadores en
inmunoensayos [68] y estudios de DNA [69-70].
Los máximos rendimientos cuánticos en la emisión QL de los ésteres de acridinio
se obtienen a pH 12-13, pero a estos pHs los ésteres de acridinio se descomponen dando
una pseudo-base no-quimioluminiscente. Por esto, el H2O2 se añade en medio ácido para
reconvertir la pseudo-base a éster de acridinio y posteriormente se añade el hidróxido
sódico para iniciar la reacción QL.
Al igual que en la reacción del luminol, también se puede generar el H2O2
electroquímicamente.
Figura 5. Mecanismo de la reacción QL de los ésteres de acridinio.
La lucigenina, nitrato de bis-N-acridinio, no lleva el grupo éster, pero sigue el
mismo mecanismo que los ésteres, con la única diferencia que requiere de la presencia de
iones metálicos de transición que actúan como catalizadores. Por esta razón, se puede
utilizar también la reacción para la determinación de iones metálicos si se utiliza
lucigenina como reactivo quimioluminiscente.
N
+
CH3
C O
O
R
OH, H2O2
N
CH3
O
*
Ester de acridinio
N-Metil acridona*
NMA*
NMA + h ν](https://image.slidesharecdn.com/meseguer-170216182421/85/Meseguer-52-320.jpg)
![Capítulo 1. INTRODUCCIÓN
29
1.3.5.3. Reacción de Tris (2,2’-bipiridina) rutenio (III)
Las propiedades fotoluminiscentes de los N’N’ quelatos de Ru(II) fueron
presentadas por primera vez en 1959, y posteriormente en 1962, se observó la QL emitida
por el Tris(2,2’-bipiridina) rutenio (III). Sin embargo, hay muy poca información acerca
del mecanismo de la reacción.
El estado excitado del complejo Ru (bipi)3
2+
, produce una emisión naranja a 610
nm. La emisión es fosforescencia químicamente inducida. Se puede llegar a este estado
excitado de diferentes formas [45]:
§ Ru(bipi)3
3+
+ Ru(bipi)3
+
→ [Ru(bipi)3
2+
]*
§ Ru(bipi)3
+
+ Oxidante → [Ru(bipi)3
2+
]* + Oxidante-
§ Ru (bipi)3
3+
+ Reductor → [Ru(bipi)3
2+
]* + Reductor+
La intensidad de quimioluminiscencia en estas reacciones es proporcional a la
concentración de cualquiera de los reactivos.
Como reductores se pueden utilizar una gran variedad de compuestos, pero no
todos producen la típica QL naranja. Algunos que sí la producen son: aminas [71-72],
aminoácidos [73], alcaloides [74-75] e ión oxalato [76-77], a concentraciones incluso por
debajo de 1· 10-10
M.
La reacción es tan selectiva que incluso para alcaloides como la codeína y la
morfina con una estructura química tan similar, la codeína se puede determinar, pero la
morfina no da QL. Esto ha dado lugar a que no existan todavía mecanismos definitivos
para este tipo de reacción. Para las aminas, la respuesta aumenta con el número de
sustituyentes (terciarias > secundarias > primarias).
Recientemente, se ha estudiado un complejo similar Ru(Phen)3
2+
(Phen: 1,10-
fenantrolina), que presenta mayor emisión al oxidarse con Ce(IV) en medio sulfúrico.
1.3.5.4. Reacciones Bioluminiscentes
Los sistemas más utilizados se basan en la reacción de luciferina-luciferasa [78-
79]. La enzima luciferasa (muy específica) cataliza la oxidación por el aire de la
luciferina en presencia de ATP, que se consume como substrato y emite luz a 562 nm.
Para esta reacción es necesaria la presencia de Mg(II) para que la luciferasa muestre su
actividad. Por cada molécula de ATP consumida, se emite un fotón, por esto, esta
reacción es un sistema analítico ideal para detectar la presencia de ATP.](https://image.slidesharecdn.com/meseguer-170216182421/85/Meseguer-53-320.jpg)
![QUIMIOLUMINISCENCIA
30
Este sistema se emplea en el ámbito de la gestión y garantía de calidad para
aumentar la higiene en alimentos, en control de calidad ambiental, en el seguimiento de
plantas de aguas residuales y en la industria farmacéutica, entre otros.
1.3.5.5. Reacciones de oxidación directa
Existen reacciones quimioluminiscentes por combinación de una especie dada
con oxidantes y reductores fuertes en diferentes condiciones químicas. Se han utilizado
estas reacciones para la determinación de aminoácidos, SO2 o morfina con MnO4
-
en
medio ácido y alcalino [80-81], ácido ascórbico con Ce(IV) [82], nitrito con H2O2 [83],
tetraciclina con Br2 [84], entre otras aplicaciones. Otros oxidantes utilizados
habitualmente son el ClO-
y el IO4
-
. Normalmente, cuando la oxidación o la reducción del
analito da un producto fluorescente o el analito en sí presenta fluorescencia, cabe la
posibilidad de que la oxidación del analito produzca emisión QL. Para aumentar la
emisión QL en estos casos, se han añadido al medio de reacción sensibilizadores, medios
micelares o catalizadores. Estas reacciones, se han aplicado principalmente a métodos de
inyección en flujo con detección quimioluminiscente.
1.3.6. REACCIONES QUIMIOLUMINISCENTES INDIRECTAS EN FASE
LÍQUIDA
1.3.6.1. Reacciones con dioxoetanos
Los dioxoetanos son peróxidos cíclicos de 4 miembros. Muchos de ellos son
inestables y se han descrito como intermediarios en muchas reacciones
quimioluminiscentes. Su estabilidad relativa depende de los grupos sustituyentes. A
temperatura ambiente, los 1,2-dioxoetanos sustituidos son estables, pero pueden
descomponerse químicamente produciendo la QL. Los 1,2-dioxoetanos utilizados en las
reacciones quimioluminiscentes tienen un grupo adamantil en una parte del anillo, y un
grupo arílico sustituido (ArilO-X) en la otra. La estabilidad de esta molécula se pierde al
añadir un reactivo que provoca una reacción de transferencia de carga eliminando el
grupo X que estabilizaba la estructura, dando lugar a su descomposición espontánea que
proporciona un éster arílico en estado excitado. El mecanismo detallado [40] se puede ver
en la Figura 6.
La emisión QL la darán, tanto la especie arílica excitada de forma directa, como
un fluoróforo añadido a la mezcla de reacción al cual le transfiere la energía (QL
indirecta). Esta reacción, se ha utilizado para la determinación de dioxoetanos como los
5-tert-butil-1-(4-hidroxibenz[d]oxazol-6-il)-4,4-dimetil-2,6,7-trioxabiciclo[3.2.0]heptanos
[85]; para la determinación de los fluoróforos, como β-dicetonatos de praseodimio (III),
neodimio (III) e Yterbio (III) excitados en la descomposición de 1,2-dioxoetano [86]; o](https://image.slidesharecdn.com/meseguer-170216182421/85/Meseguer-54-320.jpg)
![Capítulo 1. INTRODUCCIÓN
31
para la determinación del reactivo utilizado en la reacción de descomposición del 1,2-
dioxetano como el enzima proteasa [87].
Figura 6. Mecanismo de la reacción QL de dioxoetanos.
1.3.6.2. Reacciones con peroxioxalatos
En QL indirecta, el sistema más frecuentemente utilizado se basa en la reacción
quimioluminiscente de los peroxioxalatos (PO-CL). En estas reacciones, un éster aril
oxalato es oxidado por peróxido de hidrógeno en presencia de un fluoróforo. El
mecanismo propuesto [45] utilizando bis-(2,4,6-triclorofeniloxalato) (TCPO) se muestra
en la Figura 7. En la reacción, se forma un intermedio de elevada energía, la 1,2-
dioxoetanodiona, que forma un complejo de transferencia de carga con el fluoróforo; el
fluoróforo cede un electrón al intermedio y el complejo se disocia, alcanzando el
fluoróforo un estado excitado. La emisión se produce al volver el fluoróforo al estado
fundamental. Por tanto, las mejores condiciones de emisión quimioluminiscente se
obtendrán cuanto más fácilmente oxidable sea el fluoróforo.
Figura 7. Mecanismo propuesto para la reacción QL con peroxioxalato.
Cl
Cl
Cl
O C
O
C
O
O
Cl
Cl
Cl
+ H2O2
Cl
Cl
Cl
O C
O
C
O
O OH
O
C
O
C
O O
F
O
C
O
C
O O
.-
F
.+
F* + 2CO2
F + hν
O-
O
CH3O
*
O O
OCH3
OX
O O
OCH3
O-
hν](https://image.slidesharecdn.com/meseguer-170216182421/85/Meseguer-55-320.jpg)
![QUIMIOLUMINISCENCIA
32
Puede añadirse a la reacción una base débil como trietilamina o imidazol, para
catalizar la reacción y aumentar la intensidad de QL obtenida. La reacción se da en un
amplio rango de pH, desde 5 a 9, pero generalmente se utiliza un pH cercano a la
neutralidad. El principal inconveniente de la reacción es la limitada solubilidad y
estabilidad de los peroxioxalatos en medio acuoso. Generalmente se utilizan disolventes
orgánicos para evitar problemas de degradación del reactivo.
En la Tabla 8, se pueden ver las características de algunos de los ésteres de
oxalato [15] utilizados en esta reacción. La QL total es el área bajo la curva de emisión.
De estos ésteres, los más utilizados son el TCPO y el DNPO.
Tabla 8. Principales características de algunos peroxioxalatos.
Reactivo quimioluminiscente QL Total
Tiempo de
emisión (s)
Bis(2,4-dinitrofenil)oxa lato DNPO 7500 11
Bis(2,4,6-triclorofenil)oxalato TCPO 8990 23
Bis(2-(3,6,9-trioxadeciloxicarbonil)-4-
Nitrofenil)oxalato TDPO
9370 13
Bis(2,6-fluorofenil)oxalato DFPO 5650 97
Bis(2-(3-oxabutiloxicarbonil)-4-
Bromofenil)oxalato MBO-1
4630 630
Bis(2-(3,6-dioxaheptiloxilcarbonil)-4-
Bromofenil)oxalato MBO-2
5000 750
Este tipo de reacciones, se ha utilizado para la determinación cuantitativa de H2O2
obteniendo límites de detección del orden de 0.18 pM [88]. La aplicación analítica más
común, ha sido la detección en cromatografía líquida de fluoróforos o compuestos que no
son fluorescentes pero pasan a serlo al derivatizarlos con cloruro de dansilo (como
aminoácidos [89, 23], esteroides [90], aminas alifáticas [91] y ácidos carboxílicos [92]
entre otros).
Para fluoróforos fácilmente oxidables tales como los derivados del dansilo, la
detección quimioluminiscente proporciona mayor sensibilidad que la fluorescente, debido
a que no existe dispersión debida a la fuente de excitación.
En esta Tesis, la reacción con peroxioxalatos y concretamente, la reacción del
TCPO/H2O2, se ha aplicado a la determinación de Cu(II). La Coproporfirina I actúa
como fluoróforo en la reacción del peroxioxalato. En presencia de Cu(II), la intensidad
de emisión disminuye por formación del complejo Cu(II)-Coproporfirina I. Basado en
esta reacción de inhibición, se ha diseñado un sistema FIA para el screening de Cu(II) en
muestras de agua (Apéndice 6).
Además, se ha utilizado esta misma reacción para la determinación cuantitativa
y screening de NH4
+
en muestras de agua mediante HPLC. Para ello, se ha derivatizado
en disolución el amonio con cloruro de dansilo, de forma que el derivado actúa de](https://image.slidesharecdn.com/meseguer-170216182421/85/Meseguer-56-320.jpg)









![BIBLIOGRAFÍA
42
1.6. BIBLIOGRAFÍA
[1] X. Domenech, Química de la Hidrosfera. Origen y destino de los contaminantes,
Miraguano, Madrid, 1995.
[2] D. Pérez-Bendito, S. Rubio, Environmental Analytical Chemistry, Elsevier,
Amsterdam, 1999.
[3] I. Bodek, W.J. Lyman, W.F. Reehl, D.H.Roseblatt, Environmental Inorganic
Chemistry, Pergamon Press, New York, 1988.
[4] M. Radojevic, V.N. Bashkin, Practical Environmental Analysis, RSC, Cambridge,
1999.
[5] M.S. Hermann, J.Chem.Ed. 71 (1994) 323.
[6] http://www.atsdr.cdc.gov/es/toxfaqs/es_tfacts33.pdf (Agency for toxic substances and
disease registry).
[7] http://www.atsdr.cdc.gov/es/toxfaqs/es_tfacts132.html (Agency for toxic substances
and disease registry).
[8] http://www.atsdr.cdc.gov/toxprofiles/tp126-c5.pdf (Agency for toxic substances and
disease registry).
[9] Y.M. Liu, J.K. Cheng, J. of Chromatogr. A, 1003 (2003) 211.
[10] L.A.Tortajada-Genaro, P.Campíns-Falcó, Tecnología del agua, 241 (2003).
[11] M.F.Pouet, O.Thomas, B.N.Jacobsen, A.Lynggaard-Jensen, P.Quevauviller, Talanta,
50 (1999) 759.
[12] M.Valcárcel, S.Cárdenas, M.Gallego, Trends in Analytcal Chemistry, 18 (11) (1999),
685.
[13] M.Valcárcel, S.Cárdenas, M.Gallego, Trends in Analytical Chemistry, 21 (4) (2002),
251.
[14] Bruno Enríquez. Energía y tú, 14 (2001).
[15] C.Dodeigne, L.Thunus, R.Lejeune. Talanta 51 (2000) 415.
[16] H.O. Albrecht, Z. Phys. Chem. 136 (1928) 321.
[17] K. Gleu, W. Petsch, Angew Chem. 48 (1935) 57.
[18] F. McCapra, P. Richardson, Tetrahedron Lett. (1964) 3167.
[19] Encyclopaedia of Analytical Sciences. Volume 5. Academic Press Limited 1995.
[20] W.U. Palm, M. Millet, C. Zetzsch, Chemosphere, 35 (5) (1997) 1117.
[21] S.W. Tjioe, Robert J. Hurtubise, Talanta, 42 (1) (1995) 59.
[22] E. Orejuela, M. Silva, Journal of Chromatogr. A, 1007 (1-2) (2003) 197.
[23] Y. Ohba, N. Kuroda, K. Nakashima, Anal. Chim. Acta, 465 (1-2) (2002) 101.
[24] C. Molins-Legua, P. Campíns-Falcó, A. Sevillano Cabeza, Anal. Chim. Acta, 378 (1-
3) (1999) 83.
[25] K. Honda, J. Sekino, K. Mai, Anal.Chem., 55 (6) (1983) 940.
[26] I. Bronstein, B. Edwards, J.C. Voyta, J. Biolumin.Chemilumin., 4 (1) (1989) 99.
[27] T. Kinkel, H. Luebbers, E. Schmidt, P. Molz, H. J. Skrzipczyk, J. Biolumin.
Chemilumin. 4(1) (1989) 136.
[28] P. Jones, T. Williams, L. Ebdon, Anal. Chim. Acta, 237 (1990) 291.
[29] K. Tsukagoshi, Y. Obata, R. Nakajima, J. Chromatogr. A, 971 (1-2) (2002) 255.
[30] J.Z. Li, P.K. Dasgupta, Anal. Chim. Acta, 442 (1) (2001) 63.
[31] T. Pérez-Ruiz, C. Martínez-Lozano, V. Tomás, J. Martín, J. of Chromatogr. A, In
Press, Corrected Proof, Available online 4 December 2003.](https://image.slidesharecdn.com/meseguer-170216182421/85/Meseguer-66-320.jpg)
![Capítulo 1. INTRODUCCIÓN
43
[32] D. Brannegan, M. Ashraf-Khorassani, L.T. Taylor, J-Chromatogr-Sci. 39 (6) (2001)
217.
[33] K. Tsukagoshi, Y. Shikata, R. Nakajima, M. Murata, M. Maeda, Anal-Sci., 18 (11)
(2002) 1195.
[34] E. Pontén, C. Viklund, K. Irgum, S. T. Bogen, Å. N. Lindgren, Anal. Chem., 68
(1996) 4389.
[35] X.Yan, J.Chromatogr. A, 842 (1999) 267.
[36] A.M. Jiménez, M.J. Navas, Trends in Anal. Chem., 18 (5) (1999) 353.
[37] H. Shi, L.T. Taylor, E.M. Fujinari, X. Yan, J. of Chromatogr. A, 779 (1-2) (1997)
307.
[38] H. Shi, L.T. Taylor, E.M. Fujinari, J. of Chromatogr. A, 757, (1-2) (1997) 183.
[39] D.L. MacTaggart, S.O. Farwell, J.R. Burdge, Z.T. Cai, T.J. Haakenson, W.L.
Bamesberger, Atmosph. Environ., 33 (4) (1999) 625.
[40] Encyclopedia of Analytical Sciences. Volume 1. Academic Press Limited 1995.
[41] R. Escobar, Q.X. Lin, A. Guiraum, F.F. de la Rosa, Analyst, 118 (1993) 643.
[42] L.A. Tortajada-Genaro, P. Campíns-Falcó, F. Bosch-Reig, Anal. Chim. Acta, 488 (2)
(2003) 243.
[43] J. Lu, C. Lau, S. Yagisawa, K. Ohta, M. Kai, J. of Pharmac. and Biomed. Anal., 33
(5) (2003) 1033.
[44] Z. Song, C. Wang, Bioorg. & Medic. Chem., 11 (24) (2003) 5375.
[45] AM. García-Campaña, W.R.G. Baeyens, X. Zhang, F.Alés, L.Gámiz. Ars
Pharmaceutica, 42 (1) (2001) 81.
[46] D. Easwaramoorthy, Y. Yu, H. Huang, Anal. Chim. Acta, 439 (1) (2001) 95.
[47] W. Qin, Z. Zhang, Y. Peng, Anal. Chim. Acta, 407 (1-2) (2000) 81.
[48] J. Li, P.K. Dasgupta, Anal. Chim. Acta, 398 (1) (1999) 33.
[49] C. Zhang, J. Huang, Z. Zhang, M. Aizawa, Anal. Chim. Acta, 374 (1) (1998) 105.
[50] R. Wei Min, J. Nielsen, J. Villadsen, Anal. Chim. Acta, 312 (2) (1995) 149.
[51] S. Ventura, M. Silva, D. Pérez-Bendito, Anal. Chim. Acta, 266 (2) (1992) 301.
[52] L.A. Tortajada Genaro, P. Campíns Falcó, S. Meseguer Lloret, F. Bosch Reig, Anal.
Bioanal. Chem., 374 (2002) 1223.
[53] S. Meseguer Lloret, P. Campíns Falcó, L.A. Tortajada Genaro, F. Blasco Gómez,
Inter. J. Environ. Anal. Chem., 83 (5) (2003) 405.
[54] Y. Moliner Martínez, S. Meseguer Lloret, L.A. Tortajada Genaro, P. Campins Falcó,
Talanta, 60 (2-3) (2003) 257.
[55] B. Li, Z. Zhang, Sens. and Act. B: Chemical, 69 (1-2) (2000) 70.
[56] Y. Lv, Z. Zhang, F. Chen, Talanta, 59 (3) (2003) 571.
[57] H.C. Hong, H.J. Huang, Anal. Chim. Acta, 499 (1-2) (2003) 41.
[58] S. Rani Jain, E. Borowska, R. Davidsson, M. Tudorache, E. Pontén, J. Emnéus,
Biosens. and Bioelectronics, In Press, Corrected Proof, Available online 23 October
2003.
[59] O. Nozaki, H. Kawamoto, Anal. Chim. Acta, 495 (1-2) (2003) 233.
[60] G.J. Zhou, G. Wang, J.J. Xu, H.Y. Chen, Sens. and Act. B: Chemical, 81 (2-3)
(2002) 334.
[61] J.J. Li, J X. Du, J.J. Lu, Talanta, 57 (1) (2002) 53.](https://image.slidesharecdn.com/meseguer-170216182421/85/Meseguer-67-320.jpg)
![BIBLIOGRAFÍA
44
[62] K.A. Fähnrich, M. Pravda, G.G. Guilbault, Talanta, 54 (4) (2001) 531.
[63] H. Lundqvist, C. Dahlgren, Free Radical Biology and Medicine, 20 (6) (1996) 785.
[64] D. Kratky, A. Lass, P.M. Abuja, H. Esterbauer, H. Kühn, Biochim. et Biophys. Acta
(BBA) - Molecular and Cell Biology of Lipids, 1437 (1) (1999) 13.
[65] O.M. Steijger, D.A. Kamminga, A. Brummelhuis, H. Lingeman, J. of Chromatogr.
A, 799 (1-2) (1998) 57.
[66] J.M. Lin, H. Goto, M. Yamada, J. of Chromatogr. A, 844 (1-2) (1999) 341.
[67] W.J. Cooper, J.K. Moegling, R.J. Kieber, J.J. Kiddle, Mar. Chem., 70 (1-3) (2000)
191.
[68] C. Shellum, G. Gübitz, Anal. Chim. Acta, 227 (1989) 97.
[69] O. Okwumabua, B. Swaminathan, P. Edmonds, J. Wenger, J. Hogan, M. Alden,
Research in Microbiology, 143 (2) (1992) 183.
[70] K. Matsubara, S. Takeda, Y. Iwaki, J. Cicciarelli, S. Musa, J. Garcia-Gomez, Human
Immunology, 44 (1995) 37.
[71] M.E. Bolden, N.D. Danielson, J. of Chromatogr. A, 828 (1-2) (1998) 421.
[72] B.L. Waguespack, A.Lillquist, J.C. Townley, D.R. Bobbitt, Anal. Chim. Acta, 441
(2) (2001) 231.
[73] T. Pérez-Ruiz, C. Martínez-Lozano, V. Tomás, J. Martín, Talanta 58 (5) (2002) 987.
[74] N.W. Barnett, T.A. Bowser, R.D. Gerardi, B. Smith, Anal. Chim. Acta, 318 (3)
(1996) 309.
[75] G.M. Greenway, L.J. Nelstrop, S.N. Port, Analytica Chimica Acta, 405 (1-2) (2000)
43.
[76] D.R. Skotty, T.A. Nieman, J. of Chromatogr. B: Biomed. Sci. and Appl., 665 (1)
(1995) 27.
[77] N.W. Barnett, S.W. Lewis, S.D. Purcell, P. Jones, Anal. Chim. Acta, 458 (2) (2002)
291.
[78] S.I. Tu, D. Patterson, J. Uknalis, P. Irwin, Food Research International, 33 (5)
(2000) 375.
[79] J.I. Horiuchi, K. Ebie, K. Tada, M. Kobayashi, T. Kanno, Bioresource Technology,
86 (1) (2003) 95.
[80] B.J. Hindson, N.W. Barnett, Anal. Chim. Acta, 445 (1) (2001) 1.
[81] J.W. Costin, P.S. Francis, S.W. Lewis, Anal. Chim. Acta, 480 (1) (2003) 67.
[82] Y. Ma, M. Zhou, X. Jin, B. Zhang, H. Chen, N. Guo, Anal. Chim. Acta, 464 (2)
(2002) 289.
[83] C. Lu, F. Qu, J.M. Lin, M. Yamada, Anal. Chim. Acta, 474 (2002) 107.
[84] A.A. Alwarthan, A. Townshend, Anal. Chim. Acta, 205 (1988) 261.
[85] M. Matsumoto, Y. Mizoguchi, T. Motoyama, N. Watanabe, Tetrahedron Letters, 42
(50) (2001) 8869.
[86] A.I. Voloshin, N.M. Shavaleev, V.P. Kazakov, J. of Luminescence, 91 (1-2) (2000)
49.
[87] I. Bronstein, B. Edwards, C. Martin, A. Sparks, J.C. Voyta, Biotechnology Advances,
15 (3-4) (1997) 711.
[88] K. Nakashima, M. Wada, N. Kuroda, S. Akiyama, K. Imai, J. Liq. Chromatogr. 17
(10) (1994) 2111.](https://image.slidesharecdn.com/meseguer-170216182421/85/Meseguer-68-320.jpg)
![Capítulo 1. INTRODUCCIÓN
45
[89] K. Miyaguchi, K. Honda, K. Imai, J. of Chromatogr. A, 303 (1984) 173.
[90] J.H. Mike, T.J. Cleland, Anal. Chim. Acta, 259 (1) (1992) 73.
[91] M. Cobo, M. Silva, J. of Chromatogr. A, 848 (1-2) (1999) 105.
[92] T. Toyo'oka, M. Ishibashi, T. Terao, J. of Chromatography A, 627 (1-2) (1992) 75.](https://image.slidesharecdn.com/meseguer-170216182421/85/Meseguer-69-320.jpg)




![REACCIÓN DEL TCPO
50
2.1.1.1. PRECONCENTRACIÓN Y DANSILACIÓN DE AMINAS ALIFÁTICAS
UTILIZANDO CARTUCHOS C18 DE EXTRACCIÓN EN FASE SÓLIDA.
APLICACIÓN AL ANÁLISIS DE SCREENING EN MUESTRAS
MEDIOAMBIENTALES DE AGUA.
Existe muy poca información acerca de la existencia de aminas alifáticas en
aguas residuales industriales y en aguas superficiales. La cromatografía gaseosa (GC) [1-
5] y la cromatografía líquida (LC) [6-12] son las principales técnicas utilizadas para la
determinación de estos analitos. En la Tabla 10 se muestran algunos de los
procedimientos descritos en la bibliografía para la determinación de aminas en muestras
de agua a niveles de concentración bajos utilizando GC y LC.
Tabla 10. Condiciones de derivatización de aminas alifáticas.
Reactivo Muestra Tratamiento de la muestra Detec. LD Ref.
Cromatografía Gaseosa (GC)
Pentafluobenziladehido Agua
residual
Deriv. directa 15-30 min a 80 º C,
pH 10 y SPME y espaciado de
cabeza.
FID
0.4- 26
ppb
[1]
----
Agua
residual
SPME y espaciado de cabeza
(30min)
NPD
/MS
3-56 ppb [2]
---- Agua Muestras acidificadas NPD 12-40ppb [3]
---- Agua
Muestras acidificadas y espaciado
de cabeza (80ºC y 15 min).
NPD
0.2-20 ppb
0.01-3ppm
[4]
Bencenosulfonil cloruro
Agua
residual y
superficial
Deriv. directa 30 min a Tª amb.
Medio básico y extracción con
CH2Cl2.
MS 0.1 ppb [5]
Cromatografía Líquida (LC)
N-hidroxisuccinamidil
fluoresceína-O-acetato
Patrones
Deriv. 30 min 45ºC en tampón
borato pH 8.5.
FL [2]
3,5-dinitrobenzoil
cloruro (DBN)
Agua
Deriv. en C18-SPE 2 min a Tª
amb. En tampón borato pH 10.
UV 2-5 ppb [3]
Acridina-9-acetil-N-
hidroxisuccinimida
Agua
residual
Deriv. directa 10 min a 50ºC en
tampón borato pH 8-9.
FL
17-87
fmol
[4]
Cloruro de Dansilo
(DNS-Cl)
Agua
Enriquecimiento en línea en pre -
columna IRC-50. Deriv. en línea
a 65ºC en tampón borato pH 11.
CL
15-300
ppb
[5]
Fenil isotiocianato
Agua de
río, grifo,
superficial
Deriv. directa 15 min a 40ºC en
tampón carbonato. Limpieza de la
solución derivatizada (C18).
UV
0.2-0.6
ppb
[6]
4-(5’,6’-
dimetoxibenzotizoil)
fenilisotiocianato
Patrones Deriv. 30 min a 80ºC en NH4OH. FL [7]
Fluram Agua
Amberlita CG-120, recogida en
HCl, evaporada y redisuelta en 1
mL de tampón borato. Deriv. pH
10.
UV
0.02-1.2
ppb
[8]
(FMOC) Agua
Deriv. en C18-SPE. 2 min a Tª
amb. y pH 10.
FL
0.25-5
ppb
[13]
Cloruro de Dansilo
(DNS-Cl)
Agua
residual
Deriv. en C18-SPE 15 min a 85ºC
en tampón carbonato pH 9.5.
UV-
FL
3-15 ppb
Este
trabajo](https://image.slidesharecdn.com/meseguer-170216182421/85/Meseguer-74-320.jpg)
![Capítulo 2. RESULTADOS OBTENIDOS Y DISCUSIÓN
51
Debido a las propiedades químicas de las aminas alifáticas, y sus bajos niveles de
concentración en las muestras, la mayoría de los procedimientos descritos requieren
técnicas de enriquecimiento como la extracción líquido-líquido (LLE), o más
recientemente la extracción en fase sólida (SPE), o la micro extracción en fase sólida
(SPME), normalmente combinada con derivatización pre- o post-columna en los
procedimientos de HPLC. En cromatografía de gases, se ha utilizado la técnica de SPME-
espaciado de cabeza [1,2], mejorándose los límites de detección hasta en tres órdenes de
magnitud para el análisis de aminas en aire y en matrices acuosas. En HPLC, las aminas
de bajo peso molecular normalmente se extraen con resinas Amberlita CG-120 [12] o
sorbentes de intercambio catiónico [14], mientras que sus derivados pueden extraerse con
sorbentes C18 [10]. M. Cobo y col. [9] han propuesto diferentes estrategias y condiciones
combinando derivatización y extracción, que permiten la cuantificación de estos analitos
a niveles inferiores a mg/L. La derivatización en disolución ha sido aceptada como un
paso efectivo [15], sin embargo, como puede verse en la Tabla 10, existen muchos
inconvenientes como el excesivo manejo de la muestra, tiempos de reacción largos,
temperaturas elevadas o consumo de disolventes. El uso de SPE para la extracción de los
derivados [10] previamente formados en disolución, puede resolver algunos problemas, y
gracias a los elevados factores de concentración, se pueden obtener límites de detección
similares o incluso menores a los obtenidos utilizando GC-MS con una instrumentación
mucho más sencilla [5].
Sin embargo, los problemas relacionados con el proceso de derivatización todavía
permanecen (manejo de la muestra, falta de automatización, etc.). Una solución posible a
los problemas de pasos extra y de interferencias, es llevar a cabo la etapa de
derivatización en el soporte sólido [16].
En esta dirección, este grupo de investigación ha desarrollado una metodología en
la que todos los pasos de la preparación de la muestra (extracción, concentración,
derivatización y transferencia al sistema cromatográfico) se llevan a cabo en el mismo
soporte, en línea o fuera de línea. El método se basa en atrapar los analitos en el sorbente,
purificarlos con un disolvente adecuado y derivatizarlos pasando el reactivo a través de
los cartuchos. El analito y el reactivo se dejan reaccionar durante un período de tiempo, y
después el exceso de reactivo se elimina (si se requiere) pasando un disolvente adecuado
a través de los cartuchos. Por último, los derivados, se desorben y se recogen para
procesarlos posteriormente [17] o son transferirdos a la columna primaria en un sistema
en línea [18-19].
Se ha demostrado recientemente la utilidad de esta metodología utilizando el
reactivo-UV 3,5-dinitrobenzoil cloruro (DNB) y las aminas alifáticas (etilamina,
isopropilamina y dimetilamina) [7]. También se ha estudiado la derivatización asistida en
soporte sólido de algunas aminas alifáticas con 9-fluorenilmetil cloroformiato (FMOC)
[13]. Esta metodología la han aplicado recientemente D. Shangguan y col. [20] en la
determinación de aminoácidos y péptidos con FMOC y sorbente de sílice.
En este trabajo, se aplica la mencionada metodología al análisis de screening de
alquilaminas en muestras de agua utilizando el reactivo Cloruro de Dansilo (Dns-Cl)
como reactivo derivatizante. Las derivatizaciones con DnsCl se llevan a cabo](https://image.slidesharecdn.com/meseguer-170216182421/85/Meseguer-75-320.jpg)
![REACCIÓN DEL TCPO
52
habitualmente en medio acuoso-acetona saturado con carbonato sódico en tiempos de
reacción largos y a elevadas temperaturas de reacción, y en muchos casos, se requiere la
extracción de los analitos derivatizados para eliminar el reactivo en exceso. Sin embargo,
es interesante el estudio de procedimientos de dansilación simplificados debido a los
límites de detección tan bajos que se alcanzan. Estos límites de detección pueden
mejorarse porque los derivados dansilados proporcionan quimioluminiscencia con el
ácido oxálico bis(2,4,6-tricloro fenil éster) (TCPO/H2O2).
Se propone un método rápido y sencillo para la preconcentración y derivatización
seguido de su separación mediante HPLC y detección de aminas en muestras de agua
medioambientales a niveles de µg/l. Se estudian sistemáticamente 7 aminas alifáticas y
amonio, para obtener las mejores condiciones analíticas de retención y dansilación en
cartuchos C18. Se optimizan las condiciones de separación cromatográfica. Los derivados
de dansilo se registran a 333 nm con detección UV, y a λexc = 350 nm / λem = 530 nm con
detección fluorescente. Finalmente, el método se aplica a la determinación o screening de
las aminas estudiadas en muestras de agua. Más información acerca de las condiciones
experimentales empleadas, se refleja en el Apéndice 1 [21].
Derivatización en disolución
La respuesta obtenida para las diferentes aminas al derivatizarlas en disolución,
se tomó como referencia para las medidas en cartucho (conversión del 100%). El exceso
de reactivo se destruyó al calentar, por lo que no interfirió al tiempo de retención de los
analitos.
Preconcentración y derivatización en cartuchos C18
Los cartuchos Bond Elut C18, se seleccionaron para la preconcentración y
purificación de las aminas alifáticas basándonos en estudios previos [17]. No se observó
elución al pasar 1 mL de muestra ni al pasar el reactivo (Dns-Cl 12.5 mM en acetona :
tampón carbonato 33.3 mM pH 9.5 (2:3, v/v)) a través del cartucho.
Se estudiaron los parámetros de la reacción de derivatización: pH del medio,
tiempo y temperatura de reacción. Los intervalos estudiados para cada parámetro
manteniendo fijos los otros dos, así como los óptimos seleccionados, se muestran en la
Tabla 11. Al estudiar el pH de la reacción se observó que el rendimiento de la reacción
aumentaba hasta pH 9.5, a partir del cual se mantuvo constante (Figura 19 A). El estudio
del tiempo de reacción, proporcionó rendimientos similares entre 15 y 45 minutos. Un
tiempo de reacción de 60 minutos se consideró excesivo (Figura 19 B). El estudio de la
Temperatura de reacción, reveló un aumento importante del rendimiento al utilizar 85 ºC,
que se mantuvo hasta 110ºC (Figura 19 C). No se observaron productos de degradación
en las condiciones óptimas seleccionadas (pH 9.5, Tª 85ºC, Tiempo 15 min).](https://image.slidesharecdn.com/meseguer-170216182421/85/Meseguer-76-320.jpg)









![REACCIÓN DEL TCPO
62
2.1.1.2. DERIVATIZACIÓN EN LÍNEA DE AMINAS ALIFÁTICAS CON Dns -Cl
ACOPLADA A HPLC CON DETECCIÓN FLUORESCENTE.
La marcada tendencia en Química Analítica hacia el desarrollo de sistemas
completamente automatizados, requería alguna aportación en este sentido. Los
procedimientos de trabajo en línea, se basan en los mismos principios que los
procedimientos de trabajo fuera de línea. La instrumentación requerida así como el
manejo de la misma, es mínimo frente al equipamiento convencional. Normalmente, no
requieren la continua intervención del operador, lo que se traduce en una menor
variabilidad y menores errores sistemáticos.
Con esta intención, y continuando con el trabajo anterior, se propuso la
derivatización en línea de aminas alifáticas con cloruro de dansilo acoplada a HPLC con
detección fluorescente.
Condiciones de enriquecimiento, lavado y derivatización
Para la reacción de derivatización en línea de aminas alifáticas con cloruro de
dansilo acoplada a la separación cromatográfica de los derivados dansilados formados
con detección fluorescente, se utilizó el montaje mostrado en la Figura 14 del Capítulo
1-Introducción (Pág. 39). En este montaje, las muestras se inyectaban en una precolumna
empaquetada con material de relleno C18 para el enriquecimiento, lavado y
derivatización de la muestra con el reactivo Dns-Cl. Más información acerca de las
condiciones experimentales se refleja en el Apéndice 2 [22].
Se optimizaron las condiciones de retención en la precolumna así como de
derivatización, tales como: volumen de muestra, concentración del reactivo, tiempo de
reacción, temperatura de reacción, relación reactivo:tampón y características de la fase
móvil. En la Tabla 18, se muestran los intervalos estudiados y las condiciones óptimas
escogidas en función de la máxima retención de los analitos en la precolumna, así como
la máxima reactividad en cuanto a la formación del derivado dansilado.
Tabla 18. Condiciones optimizadas e intervalos estudiados.
Parámetro Intervalo Óptimo
Concentración DnsCl 2.5 mM - 12.5 mM 5 mM
Volumen muestra 150 - 400 µL 150 µL
Temperatura 20 - 80ºC 60ºC
Tiempo reacción 10 - 30 min 10 min
Relación
Reactivo:tampón
40:60 o 30:70 40:60
Fase móvil Tampón pH 12 o H2O H2O](https://image.slidesharecdn.com/meseguer-170216182421/85/Meseguer-86-320.jpg)



![REACCIÓN DEL TCPO
66
cromatográficas, en 30 minutos, se consiguió en línea la retención, derivatización y
separación cromatográfica de las 7 aminas alifáticas.
Este sistema presentó la ventaja de la completa automatización frente al sistema
presentado en el capítulo anterior (Apéndice 1, [21]). Además, las condiciones de la
reacción de derivatización fueron algo menos drásticas, con la ventaja de que la misma
precolumna podía reutilizarse inyección tras inyección. En el caso de la derivatización en
cartuchos de extracción en fase sólida, un mismo cartucho solamente podía reutilizarse
una sola vez. Se consiguió mejorar los límites de detección para Butilamina, Pentilamina
y Hexilamina, aunque para metilamina, etilamina, dimetilamina y dietilamina, los límites
de detección alcanzados fueron peores. Los estudios de precisión dieron %DER inferior
al 20% en todas las aminas.
La sensibilidad varió en función de la amina estudiada, aumentando al aumentar
la longitud de la cadena alifática.
Los estudios de recuperación de la cantidad fortificada en la muestra, dieron un
comportamiento similar al obtenido al utilizar el procedimiento fuera de línea (Apéndice
1, [21]), aumentando los porcentajes de recuperación al aumentar la longitud de la cadena
alifática. El efecto matriz habría de corregirse utilizando el MOSA para las aminas de
cadena corta. El procedimiento es aplicable al screening de aminas en muestras de agua.](https://image.slidesharecdn.com/meseguer-170216182421/85/Meseguer-90-320.jpg)
![Capítulo 2. RESULTADOS OBTENIDOS Y DISCUSIÓN
67
2.1.1.3. DETERMINACIÓN DE AMINAS ALIFÁTICAS EN AGUA MEDIANTE
HPLC CON DETECCIÓN QUIMIOLUMINISCENTE.
Las aminas alifáticas son compuestos que se encuentran a bajas concentraciones
en algunos tipos de aguas, por lo que los métodos para su análisis han de ser
suficientemente sensibles. La sensibilidad y la selectividad necesarias se pueden alcanzar
por combinación de la derivatización de los analitos con técnicas de separación
(cromatografía líquida (LC), cromatografía gaseosa (GC) o electroforesis capilar (CE)) y
un método de detección de elevada sensibilidad (métodos quimioluminiscentes (QL),
electroquímicos o espectrometría de Masas). La técnica de detección quimioluminiscente
ha demostrado ser una herramienta sensible para la medida de niveles ultra trazas, con
sistemas ópticos baratos y simples que proporcionan bajo ruido de fondo y un amplio
intervalo dinámico [23].
La quimioluminiscencia de compuestos sintéticos como la lofina, el Luminol, los
ésteres de acridinio, ésteres de perioxalato y complejos de rutenio, se ha utilizado
ampliamente. Derivados del Luminol como el N-(4-(aminobutil)N-etilisoluminol (ABEI),
NN’dissinimidilcarbonato (DSC) [24-25], 4-isotiocianatoftalhidracina (ILITC) [26], 6-
isotiocianatobenzo(g)ftalacina-1, 4(2H,3H)-diona (IPO) [27], y ácido 4-(6,7-dihidro-5,8-
dioxotiazolo (4,5-g)ftalacina-2-il) benzoico N-hidroxisuccinimida éster (TPB-suc) [28],
se han utilizado como reactivos quimioluminiscentes para la determinación de
compuestos amino.
El Tris(bipiridil)rutenio(III) [Ru(bipi)3] se ha empleado para la determinación de
una gran variedad de compuestos de nitrógeno sin necesidad de derivatizar el analito
[24,29]. El método quimioluminiscente del [Ru(bipi)3] es muy selectivo para la
determinación de aminas terciarias, pero no muy sensible comparado con otras técnicas
de determinación quimioluminiscente. Este reactivo normalmente se electrogenera, dando
lugar a quimioluminiscencia electrogenerada (ECL) [30]. A pesar del elevado potencial
teórico de la ECL, solamente ciertas reacciones han encontrado aplicación en análisis,
debido a limitaciones como la presencia de especies interferentes, su compleja
optimización y su falta de reproducibilidad [31].
En presencia de un fluoróforo, los derivados oxálicos son los emisores
quimioluminiscentes no biológicos más eficientes. Los más utilizados recientemente son
el Bis-(2,4,6.triclorofenil)oxalato (TCPO), el bis (2,4-dinitrofenil)oxalato (DNPO) y el
Bis[2-(3,6,9-trioxadeciloxicarbonilfenil]oxalato. Las principales ventajas de estos
sistemas son la eficiencia y la flexibilidad. Estos sistemas requieren la presencia de un
compuesto fluorescente, obtenido normalmente a través de la reacción de derivatización
del analito. Se han utilizado reactivos derivatizantes como el Cloruro de Dansilo (Dns-Cl)
[32-33,18] para aminas biogénicas y anfetaminas, el naftaleno-2,3-dialdehido (ADA) [34]
para aminas primarias, y la luminarina [35] para aminas primarias y secundarias,
combinados con mezclado postcolumna con los reactivos TCPO y H2O2.
En este trabajo, se estudia la generación de quimioluminiscencia post-columna
con el sistema TCPO/H2O2, para aumentar la sensibilidad del procedimiento de
dansilación previamente expuesto (Apéndice 1, [21]) para aminas de bajo peso molecular](https://image.slidesharecdn.com/meseguer-170216182421/85/Meseguer-91-320.jpg)
![REACCIÓN DEL TCPO
68
en muestras de agua medioambientales, alcanzándose niveles por debajo de los ng/mL. Se
estudian sistemáticamente seis aminas alifáticas: metilamina, etilamina, butilamina,
pentilamina, hexilamina y dietilamina. Además, se incluyen también aminas biogénicas
como putrescina, cadaverina, espermina y espermidina en el estudio de screening. Por
último, se aplica el método a la determinación de aminas en muestras de agua reales. Más
información acerca de las condiciones experimentales se refleja en el Apéndice 3 [36].
Los resultados obtenidos con detección quimioluminiscente, se comparan con los
obtenidos con detección UV y fluorescente utilizando el reactivo cloruro de dansilo [21,
22] u otros reactivos como cloruro de 3,5-dinitrobenzoilo (DNB) [7],
fluorenilmetilcloroformiato (FMOC) [13] y orto-ftaldialdehido/N-Acetil-L-Cisteína
(OPA/NAC) [37].
Condiciones de preconcentración y condiciones pre y post columna
Se utilizaron las condiciones de preconcentración y derivatización establecidas
previamente en el Apéndice 1 (Pág. 52-55). Los productos de reacción formados se
eluyeron con 0.5 mL o 1 mL de acetonitrilo. Los derivados dansilados se inyectaron en el
sistema cromatográfico y se mezclaron después de su separación, con el TCPO y el H2O2.
En cuanto a las condiciones postcolumna, los parámetros que afectaban a la
respuesta quimioluminiscente de acuerdo con los estudios previos [38-39] eran:
temperatura, pH, cantidad de agua, catalizador, disolvente, concentración de TCPO,
concentración de H2O2, volumen del tubo de mezclado, volumen de la celda de detección
y velocidades de flujo de los diferentes canales. Los parámetros establecidos para este
trabajo fueron: temperatura ambiente, acetonitrilo como disolvente, imidazol como
catalizador, velocidades de flujo inferiores a 0.6 mL/min, longitud del tubo de mezclado
mínima para el montaje, volumen de la celda de detección fijado por el instrumento de
medida, concentración de TCPO superior a 0.1 mM y relación TCPO:H2O2 cercana a 5.
Los parámetros más críticos, la concentración y el pH del Imidazol (evaluados en la
bibliografía en los rangos 0.1-5 mM y pH 5-7), dieron la máxima relación señal/ruido a
concentración 1 mM y pH 7, siendo la velocidad del flujo 1.5 mL/min.
El producto de reacción entre el TCPO y el H2O2 (el 2,4,6-triclorofenol), puede
dar lugar a efecto quenching disminuyendo la señal quimioluminiscente. Para evitar este
producto, ambos reactivos se prepararon por separado y se mezclaron en el sistema (ver
montaje en la Figura 15 del Capítulo 1-Introducción, Pág. 39). Se observó que la
sensibilidad era 5 veces superior cuando el TCPO (preparado en una mezcla
acetonitrilo:tetrahidrofurano), se mezclaba con los derivados antes que el H2O2. La
concentración de TCPO se varió entre 0.5-5mM a 0.35 mL/min, aumentando la
sensibilidad al aumentar la concentración del TCPO, pero no se observaron diferencias
entre las concentraciones 2.5 mM y 5 mM, por lo que se eligió como concentración
óptima de TCPO 2.5 mM. Al variar la concentración del H2O2, se observó que la
intensidad de QL disminuía al aumentar la concentración por encima de 5 mM, además](https://image.slidesharecdn.com/meseguer-170216182421/85/Meseguer-92-320.jpg)




![REACCIÓN DEL TCPO
74
Tabla 25. Concentración de amina fortificada y concentración de amina fortificada
encontrada (µg/L) en muestras de agua reales.
Concentración fortificada (µg/L) Concentración encontrada (µg/L)
Muestra
MA EA BA PeA HA DEA MA EA BA PeA HA DEA
20 32 40 64 15 31 50 88
S1 80 80 128 106 97 136
S2 20 32 40 40 19 26 39 51
40 40 47 51
S3
40 64 80 80 128 52 72 80 88 133
100 160 200 200 200 320 90 120 178 194 186 360
100 200 200 200 112 222 189 202S4
200 200 218 211
20 32 17 39
S5
40 65 80 31 68 110
Se encontró y cuantificó Metilamina utilizando la curva de calibrado de patrones
en agua de lago (8.8±0.3 µg/l), agua de riego (7±3 µg/l) y agua residual (6000±700 µg/l),
en la que también se encontró Pentilamina (3100±200 µg/l). En agua de grifo, se encontró
y cuantificó Dietilamina utilizando la curva de calibrado de patrones (13±3, n=3) µg/l y
utilizando la curva de adición estándar (15 µg/L, n=5), siendo los resultados similares. En
agua de mar no se detectaron ninguna de las aminas estudiadas. Las muestras de agua
residual y de agua marina, se analizaron también utilizando el procedimiento
cromatográfico descrito en el Capítulo previo (Apéndice 1, [21]), utilizando dansilación
asistida en soporte sólido y detección fluorescente. Con este procedimiento, no se detectó
ninguna amina al analizar la muestra de agua marina, al igual que en el método propuesto,
y para la muestra de agua residual los resultados fueron (6040 ± 900 µg/L, n=3) y (2960 ±
160 µg/L, n=3) para metilamina y pentilamina respectivamente. Ambos valores pueden
considerarse estadísticamente iguales (con un nivel de confianza del 95%) a los obtenidos
por el método propuesto.
El cromatograma obtenido para el análisis de screening de la muestra de agua
residual (Figura 28) utilizando la Fase Móvil 1, indicó la presencia de metilamina y
putrescina y/o pentilamina; utilizando la Fase Móvil 2, se confirmó la presencia de
pentilamina y metilamina.
Los límites de detección obtenidos utilizando el procedimiento de screening
propuesto para las aminas alifáticas y biogénicas en el agua residual, calculado como 3
veces la señal del fondo dividido por la pendiente, fueron: 0.066, 0.053, 0.015, 0.2, 0.011,
0.0075, 0.027, 0.0076, 0.011 y 0.06 mg/L para MA, EA, BA, DEA, PeA, Put, Cad, HA,
Spd y Spm respectivamente.](https://image.slidesharecdn.com/meseguer-170216182421/85/Meseguer-98-320.jpg)


![Capítulo 2. RESULTADOS OBTENIDOS Y DISCUSIÓN
77
2.1. DISMINUCIÓN DEL LÍMITE DE DETECCIÓN DE AMONIO
EN AGUA E INCREMENTO DE LA EXACTITUD DE SU
DETERMINACIÓN.
El amonio es un micronutriente en los sistemas acuosos. Tiene un papel
fundamental en el ciclo del nitrógeno. Cuando se encuentra en concentraciones elevadas,
la cantidad de nutrientes aumenta en los sistemas acuosos aumentando la actividad
biológica, que lleva a un incremento del crecimiento de algas, turbidez, olor, mal sabor y
toxicidad. El amonio puede encontrarse en aguas superficiales, subterráneas o marinas a
bajas concentraciones, del orden de 10 µg/L. En aguas residuales, puede hallarse a
elevadas concentraciones, del orden de 30 mg/L debido a los procesos de amonificación y
de reducción de nitrato.
La legislación especifica las características del método de análisis que se utilice
para estimar su concentración. Su exactitud, precisión, y límite de detección han de ser el
10% del valor paramétrico [40].
Los métodos de referencia como la detección espectrofotométrica de amonio
utilizando el reactivo de Nessler, la reacción del indofenol o el electrodo selectivo de
amonio, se han utilizado ampliamente [41]. Además, en los últimos años se han
desarrollado nuevos métodos. En la Tabla 26, se resumen algunas de las características
analíticas como el límite de detección, el intervalo dinámico lineal y la técnica empleada,
para algunos de los métodos publicados en la bibliografía.
Los métodos de inyección en flujo han sido los más destacados en los últimos
años, normalmente combinados con sistemas como colectores de flujo [47], detectores de
fluorescencia con guía de ondas [49], sistemas quimioluminiscentes [50], detección
fluorimétrica basada en la complejación de amonio con o-ftaldialdehido y tioglicolato
[53], y detector de fila de diodos con el reactivo de Nessler [44]. También se han descrito
métodos que utilizan cromatografía iónica [46,48] y electrodos selectivos de amonio
[42,43,51,54].](https://image.slidesharecdn.com/meseguer-170216182421/85/Meseguer-101-320.jpg)
![REACCIÓN DEL TCPO
78
Tabla 26. Resumen de algunos procedimientos descritos en la bibliografía para la
determinación de amonio.
Ref. Técnica
LD
(µµg/L)
Intervalo
Lineal (mg/L)
[42]
Método de lavado para la determinación en flujo de
amonio con un electrodo iónico de gases.
- 0.1 – 10
[43]
Determinación NH4
+
en flujo. Detector electrodo de
gases.
3.85 3.857e-3 – 1.2857
[44]
Sistema FIA con 3 detectores: potenciométrico,
conductimétrico y fotométrico (DAP). Reactivo de
Nessler.
-
0.05 - 0.9
[45]
Sistema FIA con pre-concentración en resina de
intercambio catiónico. Basado en la reacción de
Nessler.
3 0.05 - 0.5
[46] Cromatografía iónica con detección fila de diodos. 18 0.36– 18
[47]
Sistema FIA con colector de flujo. Detección
fluorimétrica basada en la reacción OPA/sulfito/NH4
+
y pre-concentración
0.054 9e-5 - 0.135
[48] Cromatografía iónica. 12.8 0.05 – 5
[49]
Sistema FIA con detección fluorimétrica utilizando
una guía de ondas, basado en la reacción
OPA/sulfito/NH4
+
.
1.87 0.0107 - 0.0535
[50]
Sistema FIA quimioluminiscente basado en la
reacción del luminol.
7.2 0.0535 – 5.35
[51]
Biosensor iónico amperométrico basado en un
electrodo de carbono modificado.
107 0.107-1.3375
[52]
Sistema FIA con detección amperométrica indirecta
de los iones amonio. Electrodo modificado de
Clinoptilolito.
90 0.36-18
[53]
Sistema FIA con detección fluorimétrica. Basado en la
reacción OPA/tioglicolato.
2.675e-3 – 5.35
[54] Electrodo selectivo de amonio. 0.155 0.18-1.8
El estudio de la determinación de amonio se ha abordado en dos partes, con la
intención de aportar nuevos métodos que mejorasen la sensibilidad y selectividad de los
de referencia:
§ En la primera parte, se desarrolla un método fluorimétrico para la determinación de
amonio en muestras de agua basado en la formación de un isoindol. Además se hace
un estudio comparativo con los métodos de Nessler y Electrodo Selectivo de amonio,
que se revisan previamente a su utilización.
§ En la segunda parte, se propone un método para la determinación quimioluminiscente
de amonio en aguas por HPLC. El amonio se derivatiza en disolución con el reactivo
cloruro de dansilo, y posteriormente el derivado se separa mediante HPLC y se hace
confluir post-columna con TCPO y H2O2 generando la señal quimioluminiscente.](https://image.slidesharecdn.com/meseguer-170216182421/85/Meseguer-102-320.jpg)
![Capítulo 2. RESULTADOS OBTENIDOS Y DISCUSIÓN
79
2.1.2.1. DETERMINACIÓN DE AMONIO EN MUESTRAS DE AGUA
UTILIZANDO EL REACTIVO OPA-NAC. ESTUDIO COMPARATIVO CON
LOS MÉTODOS DE NESSLER Y ELECTRODO SELECTIVO DE AMONIO.
En este capítulo, se propone un método basado en la derivatización de amonio
con el reactivo o-Ftaldialdehido (OPA) y N-Acetil-L-Cisteína (NAC) en condiciones
básicas. El derivado isoindólico formado se detectó fluorimétricamente a λexcitación = 415
nm y λemisión = 485 nm o λexcitación = 333 nm y λemisión = 462.5 nm.
Se establecieron las condiciones óptimas para obtener la máxima sensibilidad y
selectividad, y los resultados se compararon con los obtenidos utilizando el método del
reactivo de Nessler y el método del electrodo selectivo. Los tres métodos ensayados, se
examinaron quimiométricamente para evaluar la presencia o ausencia de errores
sistemáticos y mejorar sus incertidumbres al analizar muestras desconocidas. Se
compararon las ventajas y desventajas de cada uno de ellos en términos de sensibilidad,
selectividad, exactitud y precisión. Más información acerca de las condiciones
experimentales se refleja en el Apéndice 4 [55].
Método de OPA-NAC
Se observó una variación de la señal fluorescente del isoindol formado en la
reacción amonio/OPA/NAC con el tiempo de reacción, diferente en función de la
longitud de onda seleccionada. A λexc = 415 nm / λemisión = 485 nm, la señal aumentaba
con el tiempo, alcanzando un máximo a 5 minutos (óptimo seleccionado) (Figura 29 a);
por otra parte, a λexc = 333 nm / λem = 462.5 nm, la señal disminuía con el tiempo,
seleccionándose como óptimo 2 minutos (Figura 29 b). Los espectros de emisión
fluorescente para un blanco y un patrón de amonio, a λexc = 415 nm / λemisión = 485 nm (t
reacción 5 min) y λexc = 333 nm / λem = 462.5 nm (t reacción 2 minutos), se muestran en
la Figura 30 a) y b) respectivamente.
Se optimizaron las condiciones de la reacción de derivatización: concentraciones
del reactivo OPA/NAC y pH del Tampón Borato, utilizando un diseño factorial 32
(Tabla
27) a dos niveles de pH (10.6 (-) y 10.8 (+)), dos concentraciones de OPA (8.8 mM (-) y
15 mM (+)), y dos concentraciones de NAC (8.8 mM (-) y 60 mM (+)). Las condiciones
óptimas (máxima sensibilidad) seleccionadas fueron: 8.8 mM OPA:NAC (1:1) a pH 10.8.
En la Tabla 28 se dan los resultados en cuanto a la importancia e interacción entre los
factores estudiados siendo el valor Dreferencia = 14.88.](https://image.slidesharecdn.com/meseguer-170216182421/85/Meseguer-103-320.jpg)
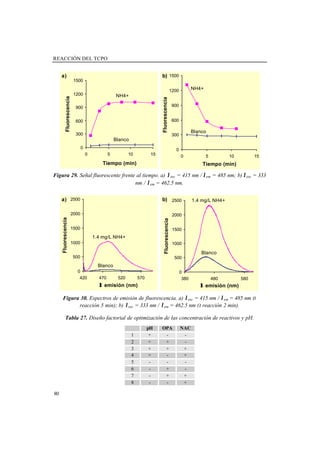
![Capítulo 2. RESULTADOS OBTENIDOS Y DISCUSIÓN
81
Tabla 28. Importancia e interacción entre los factores: pH, [OPA] y [NAC].
Factor o
interacción
Valor D Conclusión
PH 32.82 DpH > Dreferencia
OPA 58.65 DOPA > Dreferencia
NAC 976.50 DNAC > Dreferencia
Importancia de los factores:
[NAC] > [OPA] > pH
PH/OPA 11.26 DpH/OPA < Dreferencia No hay interacción
PH/NAC 32.17 DpH/NAC > Dreferencia
OPA/NAC 58.34 DOPA/NAC > Dreferencia
Dos interacciones importantes:
OPA/NAC > pH/NAC
Las curvas de calibrado de amonio a las diferentes λexc y diferentes tiempos de
reacción, y otros parámetros analíticos como límites de detección, límites de
cuantificación y desviación estándar del procedimiento, se muestran en la Tabla 29.
Como la reactividad del OPA-NAC con aminas primarias es bien conocida, se
estudió su interferencia en la determinación de amonio a las dos longitudes de onda de
excitación. A λexc = 415 nm, MA, EA, IPA y β-FEA no dieron ninguna señal fluorescente
significativa, incluso a concentraciones del orden de 15 y 30 mg/l. La curva de calibrado
de amonio en presencia de 1 mg/L de MA (Tabla 29) corrobora este hecho, manteniendo
tanto la pendiente como la ordenada igual a la curva de calibrado de patrones sin MA. Por
otra parte, en la Tabla 29 se observa que a λexc = 333 nm, la pendiente de la curva de
calibrado en presencia de MA, fue similar a la obtenida con patrones sin MA; no así
ocurrió para la ordenada, lo que indica que a esta λexc, la metilamina está contribuyendo a
la señal obtenida.
Tabla 29. Curvas de calibrado de amonio y parámetros analíticos utilizando el reactivo
OPA-NAC.
Condiciones
t
(min)
a ±± sa
b ±± sb
(n, r
2
, sy/x)
DL
(mg/L)
QL
(mg/L)
SDP
(sy/x/b)
OPA/NAC 8.8mM
(1:1), λexc = 415 nm
5 48 ± 11
759 ± 7
(10, 0.9983, 17)
0.07 0.2 0.02
OPA/NAC 8.8mM
(1:1), λexc = 333 nm
5 610 ± 30
360 ± 40
(6, 0.943, 60)
0.5 1.6 0.16
OPA/NAC 8.8mM
(1:1), λexc = 333 nm
2 450 ± 50
1380 ± 70
(6, 0.9899, 90)
0.19 0.6 0.06
* OPA/NAC 8.8mM
(1:1), λexc = 415 nm
5 60 ± 11
737 ± 16
(5, 0.9985, 18)
0.07 0.2 0.02
* OPA/NAC 8.8mM
(1:1), λexc = 333 nm
2 1380 ± 110
1380 ± 160
(5, 0.962, 180)
- - 0.13
* Calibrado de amonio en presencia de 1 mg/l de MA.](https://image.slidesharecdn.com/meseguer-170216182421/85/Meseguer-105-320.jpg)
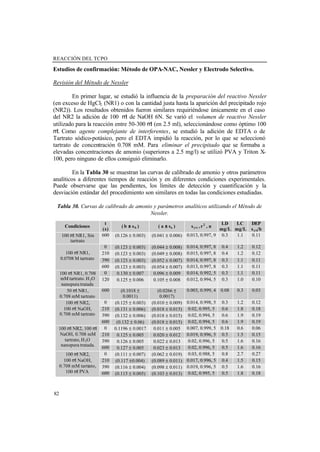

![REACCIÓN DEL TCPO
84
Análisis de muestras de agua
Aplicación del Método de Nessler
Mediante un test t (á = 0.05) se comprobó que el tratamiento de decloración y
precipitación de los cationes de las muestras, no afectaba a los resultados, siendo la
pendiente de la curva realizada con patrones tratados, similar a la pendiente de la curva de
patrones sin tratamiento (ver en Tabla 30).
Para evaluar la presencia o ausencia de errores sistemáticos, se aplicaron el
MOSA y el Youden a tres muestras de agua. Al aplicar el MOSA, las pendientes fueron
en todos los casos estadísticamente diferentes a la pendiente de la curva de calibrado de
patrones. Para las tres muestras, las pendientes variaban en función del volumen de
muestra estudiado y del tratamiento aplicado. Este comportamiento se refleja en la
Figura 33.
Figura 33. Pendientes de las curvas de calibrado al aplicar el Método de Nessler en las
diferentes condiciones estudiadas. (♦) Agua de riego (S1) ( ) Agua residual (S2) ( ) Agua
de fuente (S3). Condiciones: NR1 o NR2 (Reactivo de Nessler 1 o 2); T: Muestra Tratada;
NT: Muestra no tratada; TNC: Muestra tratada sin declorar; V: Volumen de muestra (mL);
Volumen total= 2.5 mL.
El estudio del error sistemático constante mediante el método de Youden [56],
dio una ordenada en el origen diferente de cero para la muestra de agua residual (S2)
desde t = 210 s a t = 390 s, por lo que el valor del TYB (Blanco Total de Youden) se tuvo
en cuenta al calcular la concentración de amonio en la muestra. La Figura 34, muestra
este resultado.
0
0.1
0.2
0.3
0.4
0.5
0.6
NT,V=2.4
TNC,V=2.4
T,V=2.4
T,V=2.4
T,V=1.5
T,V=1.5
T,V=0.05
NT,V=0.05
T,V=0.1
T,V=0.1
T,V=0.2
T,V=2.2
Condiciones
(b±sb)
NR1 NR2
Valor de
Referencia](https://image.slidesharecdn.com/meseguer-170216182421/85/Meseguer-108-320.jpg)
![Capítulo 2. RESULTADOS OBTENIDOS Y DISCUSIÓN
85
Figura 34. Gráfico de Youden a diferentes tiempos para la muestra de agua residual.
De estos estudios, se concluyó que al utilizar el Método del Reactivo Nessler para
calcular la concentración de amonio en muestras de agua reales, habría de aplicarse el
Método de Adición Estándar (MOSA) teniendo en cuenta el valor del TYB cuando fuese
necesario, o aplicar el GHPSAM [57], que permite determinar la concentración del
analito en muestras desconocidas eliminando ambos errores sistemáticos a la vez.
Las concentraciones de amonio encontradas en las tres muestras estudiadas
utilizando este método aplicando el MOSA o el GHPSAM, se muestran en la Tabla 31.
Estas concentraciones responden a las concentraciones habituales de amonio en estos
tipos de muestras.
Aplicación del Método de Electrodo Selectivo
En la bibliografía [58] se recomendaba aplicar el método de adición estándar si la
muestra presentaba cantidades muy bajas de amonio, por lo que se aplicó el MOSA a la
muestra de agua de riego. La pendiente del MOSA (b ± sb) = (-56.3 ± 0.5), resultó
estadísticamente igual a la pendiente de la curva de calibrado con patrones (b ± sb) = (-6.6
± 1.2). El cálculo de la concentración de amonio en las muestras, se realizó por medida
directa de la señal e interpolación en la curva de calibrado de patrones. Los resultados se
muestran en la Tabla 31.
-0.2
-0.1
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
0 0.05 0.1 0.15 0.2 0.25
Volumen de muestra (ml)
Absorbancia
t=0s
t=390s
t=300s
t=210s
t=90s](https://image.slidesharecdn.com/meseguer-170216182421/85/Meseguer-109-320.jpg)



![Capítulo 2. RESULTADOS OBTENIDOS Y DISCUSIÓN
89
2.1.2.2. DETERMINACIÓN QUIMIOLUMINISCENTE DE AMONIO EN AGUAS
POR HPLC.
Los métodos de referencia para la determinación de amonio, Nessler, el indofenol
y electrodo selectivo, presentan problemas de selectividad [55,59-61]. En la bibliografía
se han descrito algunos procedimientos para mejorarla. En estos trabajos, se ha utilizado
una resina de intercambio aniónico ligeramente básica [62] o bien una columna de
intercambio catiónico de alta capacidad [63], para separar el amonio de otros cationes
inorgánicos y/o aminas alifáticas que pueden presentar las muestras de agua.
Existen también métodos para medir amonio basados en las reacciones de
derivatización con o-ftaldialdehido (OPA), como el reactivo OPA-sulfito [59] o más
recientemente el OPA-tioglicolato [64] y el OPA-NAC (descrito en el capítulo
precedente, Apéndice 4 [55]) por formación de derivados fluorescentes, que si bien
presentan mejores prestaciones que los anteriores, pueden conducir a errores sistemáticos
en algunas ocasiones, ya que la fluorescencia del fondo debida a compuestos orgánicos
naturales no suele ser insignificante en aguas naturales [59].
También se han desarrollado procedimientos quimioluminiscentes para la
determinación de amonio [65-66] debido a su elevada sensibilidad, rapidez, simplicidad y
viabilidad. El reactivo utilizado normalmente es el luminol combinado con el hipoclorito,
en sistemas de inyección en flujo, pero no ha sido descrita su aplicación a HPLC para
mejorar su selectividad.
Aunque la reacción entre los compuestos de nitrógeno y el cloruro de dansilo
(DnsCl) es bastante conocida, no se ha aplicado a la determinación cuantitativa de iones
amonio con sistema de detección fluorimétrico o con generación quimioluminiscente
post-columna (con reactivos como Bis-(2,4,6-triclorofenil)oxalato, TCPO). Sin embargo,
la reacción de dansilación se ha estudiado ampliamente para la determinación de analitos
como aminas biogénicas en plantas [67], aminas de bajo peso molecular en muestras de
agua como se ha descrito en capítulos precedentes [68,21,36], fenoles en aguas
superficiales [69], residuos de pesticidas del tipo fenoxil N-metilcarbamato en zumos de
frutas [70] y anfetaminas en muestras de orina [71,18].
Basándonos en la experiencia de nuestro grupo de investigación en la
derivatización de aminas con Cloruro de Dansilo y TCPO [36,71,18], se desarrolla en este
trabajo un método quimioluminiscente rápido, sensible y selectivo para el screening y
cuantificación de amonio en muestras de agua de baja concentración de amonio (<0.5
mg/l), pero también aplicable a muestras de concentración más elevada.
El método se basa en la derivatización directa de amonio en la muestra (sin
ningún tratamiento previo) con el reactivo cloruro de dansilo, seguido de HPLC para
separar la señal analítica del amonio de la respuesta de otros compuestos de nitrógeno.
Después de la separación en una columna analítica C18, los derivados se mezclaron con
el TCPO/H2O2 y se registró la señal quimioluminiscente.](https://image.slidesharecdn.com/meseguer-170216182421/85/Meseguer-113-320.jpg)
![REACCIÓN DEL TCPO
90
Se estudió la interferencia de los derivados de aminas en la señal del derivado de
amonio, y también se propuso un procedimiento para el screening de aminas alifáticas y
amonio en muestras de agua de diferente naturaleza. Además, el método se aplicó a varias
muestras después de aplicar el tratamiento Kjeldahl, para obtener valores de Nitrógeno
Kjeldahl Total.
Más información acerca de las condiciones experimentales se puede encontrar en
el Apéndice 5.
Optimización de la dansilación precolumna del amonio
La dansilación convencional de aminas se basa en la reacción directa con cloruro
de dansilo en presencia de una concentración elevada de bicarbonato, y algunos autores
recomiendan que la reacción transcurra durante toda la noche (12 horas a temperatura
ambiente). En este trabajo, el amonio se derivatizó de acuerdo con las condiciones
establecidas por Marcé y col. para poliaminas [67], con algunas modificaciones
introducidas para hacer más rápido el procedimiento. Para establecer las condiciones
óptimas, los derivados dansilados se inyectaron en el sistema cromatográfico, y la
reacción quimioluminiscente post-columna se llevó a cabo siguiendo el procedimiento
descrito por nuestro grupo de investigación para aminas alifáticas previamente
desarrollado [36] (ver el montaje utilizado en la Figura 15, Pág. 39 del Capítulo 1-
Introducción). El derivado dansilado de amonio eluyó a un tiempo de retención de 2.4
minutos (Figura 35).
El cromatograma del blanco presentó un pico al mismo tiempo de retención que
el pico del amonio. Este pico aparecía en mayor o menor medida en función de la
cantidad de disolvente empleado (Acetonitrilo 1, Scharlau) en la disolución del reactivo.
Se intentó separar este pico del pico del derivado de amonio, mediante la utilización de
diferentes gradientes de elución y también utilizando una precolumna con diferentes tipos
de relleno (CN, C18, SCX, o SAX) insertada mediante una válvula de conmutación, pero
en ningún caso se obtuvo separación, lo que indicaba que el pico se debía a una
contaminación de amonio en el disolvente empleado.
Por ello, se ensayaron diferentes disolventes: acetona, acetonitrilo 2 (J.T.Baker),
tetrahidrofurano y metanol. La mezcla del reactivo contenía 0.9 mL de disolvente (0.72
mL más 0.18 mL de disolución de reactivo DnsCl 5 mM), 0.2 mL de tampón carbonato
0.1 M de pH 9 y 1 mL de agua nanopura o patrón de amonio.
Aunque con disolventes como tetrahidrofurano (THF) o metanol, no apareció
pico de blanco, ninguno de ellos fue apropiado. El metanol disminuyó mucho la
eficiencia de la reacción post-columna, y el THF impidió la reacción de dansilación al
encontrarse el reactivo DnsCl y el NH4
+
en dos fases diferentes. La acetona presentó el
mismo comportamiento que el acetonitrilo 1, siendo apropiada para llevar a cabo ambas
reacciones (pre- y post- columna), pero presentaba un pico de blanco significativo. Al](https://image.slidesharecdn.com/meseguer-170216182421/85/Meseguer-114-320.jpg)

![REACCIÓN DEL TCPO
92
Figura 36. Altura de pico del Blanco frente a Volumen de Acetonitrilo (Concentración de
DnsCl fija 0.45 mM) o frente a concentración de DnsCl (Volumen de acetonitrilo fijo 0.4 mL).
De acuerdo con estos resultados, las condiciones óptimas seleccionadas fueron las
que se muestran en la Tabla 34: 0.4 mL de cloruro de dansilo 1.5 mM disuelto en
Acetonitrilo (J.T.Baker), 0.05 mL de tampón carbonato 0.2 M pH 9 y 1.55 mL de patrón
de amonio/muestra. En esta Tabla, se comparan las condiciones establecidas por Marcé y
col. [67] con las optimizadas en este trabajo. Como puede verse, se simplificó el
procedimiento original para hacerlo más rápido.
Tabla 34. Comparación del procedimiento de derivatización en disolución establecido por
Marcé y col.[67] y el procedimiento de derivatización en disolución optimizado.
Procedimiento de Marcé y colaboradores Procedimiento optimizado en este trabajo
200 ìL muestra/patrón + 40 ìL patrón
interno + 200 ìL de carbonato sódico
saturado + 400 ìL DnsCl 5 mg/ml en acetona
1.55 mL muestra/patrón + 50 ìL de tampón
carbonato 0.2 M pH 9 + 400 ìL DnsCl 1.5 mM
en acetonitrilo
Incubar en la oscuridad toda la noche a Tª
ambiente o calentar 10 min a 70 ºC
Calentar 10 minutos a 70ºC
Añadir 100 ìL de Prolina de 100 mg/ml e
incubar en la oscuridad 30 minutos
Inyectar 20 ìL en el sistema cromatográfico
Extraer los derivados dansilados en 500 ìL
de tolueno y mezclar 30 segundos
Separar 400 ìL de la fase orgánica, secar y
redisolver en 800 ìL de acetonitrilo
Pasar a través de un filtro de tamaño de poro
0.45 ìm
Inyectar 20 ìL en el sistema cromatográfico
0
10000
20000
30000
40000
50000
60000
70000
80000
Volumen de Acetonitrilo (mL) o
Concentración de DnsCl (mM)
AlturadePicodelBlanco(V)
Volumen de acetonitrilo Variable
Concentración de DnsCl Variable
Condiciones óptimas0.18 mL
0.4 mL
0.65 mL
0.9 mL
0.15 mM 0.3 mM
0.75 mM
0.45 mM](https://image.slidesharecdn.com/meseguer-170216182421/85/Meseguer-116-320.jpg)

![Capítulo 2. RESULTADOS OBTENIDOS Y DISCUSIÓN
95
recuperación de la señal del amonio fue del 89.3%, por lo que se consideró que no
afectaba su presencia a la determinación de amonio aún encontrándose a niveles
superiores a los que estas se presentan en aguas.
Análisis de muestras de agua
Se analizaron 10 muestras de agua reales, y en varias de ellas se encontró amonio.
A modo de ejemplo, en la Figura 38, se muestran los cromatogramas por triplicado
obtenidos para la muestras de agua residual (dilución 1:130) (A) y de lago (dilución
1:8.25) (B). Ambas presentaban amonio en diferentes concentraciones.
Figura 38. Cromatogramas de las muestras de agua (3 réplicas). A) Agua residual.
B) Agua de lago.
Para validar la exactitud del método se fortificaron las muestras con
concentraciones de amonio conocidas. Al aplicar el MOSA, las muestras de agua
natural(S1), de grifo (S2), de fuente (S3), de riego 2 (S5), de lago(S6) y residual (S10),
presentaron pendientes similares a las de la curva de calibrado con patrones (ver Tabla
35) a un nivel de confianza del 95% como lo demostró el test t [72] dado en la Tabla 37,
por lo que no había efecto matriz y la concentración de amonio en las muestras podía
calcularse a partir de la curva de calibrado de patrones con resultados exactos. Para la
muestra S4 (agua de riego 1) la pendiente de la curva de calibrado del MOSA fue similar
a la de la curva de calibrado de patrones a un nivel de confianza del 98%. Para las
muestras de agua marina, solo S7 dio pendientes similares a un nivel de confianza del
99%, mientras que S8 y S9 presentaron efecto matriz como puede verse en la Tabla 37.
En presencia de efecto matriz, la concentración de amonio debe calcularse aplicando el
0 1 2 3 4
Tiempo (min)
SeñalQuimioluminiscente
NH4+
NH4+
A)
B)](https://image.slidesharecdn.com/meseguer-170216182421/85/Meseguer-119-320.jpg)


![REACCIÓN DEL TCPO
98
muestras utilizando la curva de calibrado de patrones de amonio (Tabla 35) y utilizando
el MOSA, observándose resultados similares. El agua de fuente y el agua residual se
analizaron también utilizando el método de OPA-NAC anteriormente descrito (Apéndice
4, [55]). Los resultados fueron 0.37 (s=0.02, n=3) y 32 (s=0.4, n=3), respectivamente.
Ambos valores pueden considerarse estadísticamente similares (a un nivel de confianza
del 95%) a los obtenidos por el método propuesto. El amonio cuantificado en el agua
residual debería observarse con reservas, debido a la gran dilución llevada a cabo en la
muestra tanto por el método quimioluminiscente como por el fluorescente.
Un grupo representativo de estas muestras se trató para evaluar el Nitrógeno
Kjeldahl Total. El test t presentado en la Tabla 37, indicó que la pendiente del MOSA era
similar a la de la curva de patrones de amonio. Por tanto, ninguna de las muestras
presentaba efecto matriz, pudiéndose obtener la concentración de amonio a partir de la
curva de calibrado con patrones. Para validar la exactitud, las muestras tratadas mediante
el procedimiento Kjeldahl, se fortificaron con concentraciones conocidas de amonio, y las
concentraciones de amonio halladas se muestran en la Tabla 38. Como puede observarse,
se obtuvieron errores relativos bajos, entre el 0.3 y el 12.7%.
La concentración de amonio en las muestras después de aplicar el tratamiento
Kjeldahl, fue superior en todos los casos a la concentración del amonio libre (Tabla 39).
Las muestras de agua de fuente, residual y de lago, dieron una respuesta de screening
positiva, siendo su concentración superior al límite establecido por la legislación (2
mg/L).
Tabla 39. Respuesta de screening y Concentración de amonio deducida para las muestras
estudiadas.
Muestra
Límite
legislado
(mg/l)
Respuesta
Screening
Concentración NH4
+
(C ±± s) (mg/l) (n=3)
Curva patrones
Concentración NH4
+
(C ±± s) (mg/l) (n)
MOSA
S1-Natural 0.5 No - -
S2-Grifo 0.5 No - -
S3-Fuente 0.5 No (0.367 ± 0.012) 0.347 (n=5)
S4-Riego 1 0.5 No - -
S5-Riego 2 0.5 No - -
S6-Lago 0.5 No (0.352 ± 0.014) 0.378 (n=5)
S7-Marina 1 0.5 Si (3.8 ± 0.1) 3.108 (n=5)
S8-Marina 2 0.5 No Efecto matriz 0.098 (n=5)
S9-Marina 3 0.5 No Efecto matriz 0.147 (n=5)
S10-Residual 0.5 Si (30 ± 4) 30.69 (n=4)
S3 Kjeldahl 2 Si (2.28 ± 0.12) 2.34 (n=3)
S4 Kjeldahl 2 No (0.56 ± 0.10) 0.595 (n=4)
S6 Kjeldahl 2 Si (3.05 ± 0.13) 2.71 (n=3)
S9 Kjeldahl 2 No (0.81 ± 0.08) 0.885 (n=5)
S10 Kjeldahl 2 Si (169 ± 18) 152.6 (n=4)](https://image.slidesharecdn.com/meseguer-170216182421/85/Meseguer-122-320.jpg)

![REACCIÓN DEL TCPO
100
2.2. MÉTODO DE SCREENING DE MUESTRAS PARA Cu(II).
La necesidad de respuestas analíticas rápidas y fiables es incuestionable. Los
métodos de screening de muestras [73-74] proporcionan una respuesta binaria Si/No, que
indica si los analitos en cuestión están presentes en la muestra a un nivel de concentración
por encima o por debajo de un nivel de corte establecido. Con esto, se consiguen algunas
ventajas, como la reducción de los costes, rapidez, simplicidad y minimización de errores
debido a las diferencias entre el muestreo y el análisis.
Los métodos de screening tienden a dar información cualitativa más que
cuantitativa, requieren muy poco o ningún tratamiento de la muestra, son rápidos y
normalmente requieren confirmación mediante el uso del método convencional para las
muestras cuyo resultado ha sido positivo. Ha de establecerse el límite de detección, el
valor de corte alrededor del cual se establecerán las posibilidades de error del método, el
umbral o valor establecido por el cliente o la legislación (marcará la respuesta Sí/No), y
las incertidumbres de los valores anteriores.](https://image.slidesharecdn.com/meseguer-170216182421/85/Meseguer-124-320.jpg)
![Capítulo 2. RESULTADOS OBTENIDOS Y DISCUSIÓN
101
2.1.3.1. MÉTODO AUTOMÁTICO DE INYECCIÓN EN FLUJO PARA EL
SCREENING DE MUESTRAS DE AGUA PARA Cu(II), BASADO EN LA
REACCIÓN QUIMIOLUMINISCENTE DE COPROPORFIRINA I-Cu (II) /
TCPO / H2O2.
Igarashi y col. [75] propusieron un método para la determinación de Cu(II)
basado en la reacción quimioluminiscente de la Coproporfirina I-Cu(II)/TCPO/H2O2 en
estático. Basándonos en estos estudios, se propone en este capítulo un método automático
para el screening de muestras de agua para Cu (II), basado en la reacción
quimioluminiscente del peroxioxalato. La Coproporfirina I es el fluoróforo. En presencia
de Cu(II), por efecto quenching, la emisión quimioluminiscente disminuye. Se utilizó un
detector quimioluminiscente simple diseñado para inyección en flujo.
Se analizaron varias muestras de agua y se validó el método mediante el análisis
de un patrón de referencia certificado, verificándose su utilidad para cumplir con la
legislación vigente sobre Cu (ver Tabla 3, Pág. 14 del Capítulo1-Introducción)
El montaje de inyección en flujo utilizado es el que se muestra en la Figura 16
Pág. 40 del Capítulo 1-Introducción. Más información acerca de las condiciones
experimentales se incluye en el Apéndice 6.
Estudio de la señal del Detectorde Quimioluminiscencia FireFly
Se evaluó la sensibilidad del detector utilizando el procedimiento en flujo descrito
para la reacción del Cr(III)/Luminol/H2O2 [76]. Las curvas de calibrado Log-Log y los
límites de detección obtenidos por nuestro grupo de investigación en publicaciones
recientes [76-78] utilizando el espectrofluorímetro Hitachi F-4500, así como la curva
obtenida con este detector, se muestra en la Tabla 40. El detector de
Quimioluminiscencia FireFly presentó sensibilidad similar al Hitachi F-4500, con la
ventaja de ser mucho más simple y presentar mucho menor coste.
Tabla 40. Comparación de sensibilidad de los detectores en la reacción QL
Cr(III)/luminol/H2O2.
Referencia (loga ±± sloga) (b ±± sb)
Límite de
detección (µµg/l)
Detector
[18] (1.91 ± 0.03) (1.19 ± 0.03) 1.3 Hitachi F4500
[19] (1.97 ± 0.07) (1.25 ± 0.07) 1.2 Hitachi F4500
[20] (1.84 ± 0.03) (1.16 ± 0.03) 1.6 Hitachi F4500
Este trabajo (1.04 ± 0.06) (1.25 ± 0.06) 1.4 FireFly QL](https://image.slidesharecdn.com/meseguer-170216182421/85/Meseguer-125-320.jpg)

![Capítulo 2. RESULTADOS OBTENIDOS Y DISCUSIÓN
103
En cuanto a las variables químicas estudiadas, destacar que la presencia de CTAC
sensibilizó la reacción por ofrecer un entorno más adecuado. El H2O2 y el TCPO debían
llegar al detector por separado para alcanzar la máxima sensibilidad. Sus concentraciones
se variaron de acuerdo con los estudios recientes de esta reacción quimioluminiscente
[79-80]. El TCPO se preparó en acetonitrilo:tetrahidrofurano 75:25 para evitar su
degradación y obtener la máxima sensibilidad. El pH óptimo para la reacción se obtuvo
con tampón bórax 0.1 M a pH 7.4.
En cuanto a las variables físicas, del estudio de la velocidad de la bomba 2, se fijó
una velocidad de flujo para el TCPO de 2.9 ml/min, y para la Coproporfirina I, de 5
ml/min. El volumen del bucle seleccionado fue de 50 ìL. Volúmenes mayores dieron
problemas de reproducibilidad y presión. Dada la rapidez de la reacción, parámetros
como la distancia desde las válvulas de inyección al detector o la longitud de la guía de
ondas fueron estudiados. En ambos casos, los valores óptimos fueron las mínimas
distancias posibles. La sensibilidad aumentó 10 veces al acortar la distancia de las
válvulas de inyección al detector, además de evitarse problemas de dispersión del bolo de
muestra. Por su parte, al disminuir la longitud de la guía de ondas, la sensibilidad
aumentó 2.5 veces.
Con todas estas variables optimizadas, la concentración de Coproporfirina I
empleada para la reacción con Cu(II) fue 1· 10-6
M, concentraciones inferiores no dieron
reacción. Se incorporó el Cu(II) al sistema y se estudió el tiempo de reacción Cu(II)-
Coproporfirina I en el baño de agua. El efecto quenching aumentaba al aumentar en
tiempo de reacción, seleccionándose como óptimo 5 minutos.
Sensibilidad y selectividad del método
Se obtuvieron las curvas de calibrado de Cu(II) en diferentes condiciones de
velocidad de flujo de los portadores de Coproporfirina I y CTAC/H2O2 (0.7/0.5 o 2.2/0.7)
y diferentes condiciones de tiempos de reacción en los baños, t1 y t2 (min). Los resultados
se muestran en la Tabla 42.
Tabla 42. Curvas de calibrado de Cu(II) y parámetros analíticos en diferentes condiciones
(IL: Intervalo lineal; LD: Límite de detección).
vbomba1 (ml/min)
(porph. /
CTAC-H2O2)
t1 t2 a ±± sa b ±± sb
IL
(µµg/l)
LD
(µµg/l)
n, r2
, sy/x /b
0.7/0.5 5 2 4300 ± 40 -18.3 ± 0.7 0-125 17 12, 0.984, 5.38
0.7/0.5 5 2 4370 ± 110 -18.9 ± 1.3 0-125 17 8, 0.972, 8.96
0.7/0.5 5 5 6770 ± 30 -30.7 ± 0.4 0-125 10 5, 0.999, 1.32
2.2/0.7 4 0 3840 ± 20 -19.0 ± 1.0 0-32 17 4, 0.994, 1.22
2.2/0.7 5 0 4255 ± 18 -36.6 ± 1.0 0-32 8.7 4, 0.999, 0.58
2.2/0.7 5 0 5010 ± 50 -30.7 ± 0.8 0-100 10 10, 0.994, 2.83](https://image.slidesharecdn.com/meseguer-170216182421/85/Meseguer-127-320.jpg)







![Capítulo 2. RESULTADOS OBTENIDOS Y DISCUSIÓN
111
Tabla 45. Estudios recientes basados en la reacción del luminol-H2O2 para cromo.
Ref. Método
Límite Detección
(µµg/L)
[81]
Determinación de Cr(III) y Co. Separación
cromatográfica.
15
[82]
Determinación Cr(III) Inyección en Flujo. EDTA como
agente complejante.
0.01
[83]
Determinación de Cr(VI) y Cr(III). SO2 como reductor.
Separación cromatográfica.
Cr(III): 50
Cr(VI): 100
[84]
Determinación de Cr(VI) y Cr(III). Inyección en flujo.
H2O2 en HCl como agente reductor. EDTA como agente
complejante.
0.01
[85]
Determinación de Cr(III) y Cr(VI) por cromatografía
iónica. Sulfito sódico como agente reductor.
Cr(III):120
Cr(VI):90
[86] Determinación de Cr(III). Inyección en flujo. 500
[87]
Determinación Cr(VI) y Cr(III) con separación
cromatográfica y una segunda columna cromatográfica
para la reducción de Cr(VI) a Cr(III).
2
[88] Determinación de Cr(III). Inyección en flujo. 5.2
El montaje de inyección en flujo utilizado es el que se muestra en la Figura 17
Pág. 40 del Capítulo 1-Introducción. Más información acerca de las condiciones
experimentales se refleja en el Apéndice 7 [77].
Modelos de Calibración para Cr(III), Cr(VI) y Cr Total
Las curvas de calibrado Señal-Concentración, siguen una ecuación potencial del
tipo: S = a· Cb
, que al tomar logaritmos se transforma en una recta del tipo: LogS = Loga
+ b· LogC. También se han obtenido las ecuaciones de las rectas de calibrado
considerando únicamente el intervalo donde el comportamiento es lineal.
Se ensayaron reactivos de diferentes calidades: HCl(p.a.) / NaHCO3(r.a.) /
Na2CO3· 10H2O(p.a.), o bien HCl(trace pure) / Na2CO3(p.a.), y se observó que la señal del
blanco desaparecía al utilizar los reactivos de mayor pureza: HCl(trace pure) /
Na2CO3(p.a.).
En la Tabla 46, se muestran las curvas de calibrado Log-Log e intervalo lineal
para el Cr(III) y para el Cr(VI) utilizando reactivos para análisis de trazas, y en el caso
del Cr(VI), a dos niveles de HCl diferentes (1· 10-3
M y 5· 10-3
M). Al observar los
intervalos lineales, se dedujo que a mayor concentración de HCl, la sensibilidad era
mayor, lo que significa que la concentración de HCl utilizada en la reducción de Cr(VI) a
Cr(III) es un parámetro a controlar.](https://image.slidesharecdn.com/meseguer-170216182421/85/Meseguer-135-320.jpg)
![REACCIÓN DEL LUMINOL
112
Tabla 46. Curvas de calibrado para Cr(III) y Cr(VI).
Curva de calibrado
Cr HCl (M)
Log-Log:
a ±± sa; b ±± sb
(n, sy/x, r
2
)
Intervalo lineal:
a ± sa; b ± sb
(n, sy/x, r
2
)
Cr(III)* -
1.06±0.07; 1.70±0.07
(10, 0.04, 0.9861)
-330±70; 97±6
(10, 80, 0.9689)
Cr(VI) 1· 10
-3 1.576±0.012; 1.403±0.013
(40, 0.02, 0.9967)
-189±8; 115.9±1.1
(30, 17, 0.9976)
Cr(VI) 5· 10
-3 1.927±0.008; 1.314±0.009
(40, 0.014, 0.9984)
-252±17; 197±2
(30, 40, 0.996)
En la Figura 43 se muestran los picos de inyección en flujo obtenidos para el
calibrado de Cr(VI) con los reactivos de mayor pureza (HCl(trace pure) / Na2CO3(p.a.)),
y concentración de HCl 5· 10-3
M para el tratamiento de reducción de Cr(VI) a Cr(III).
Figura 43. Registros de señal para la curva de calibrado de Cr(VI) con HCl 5· 10
-3
M.
Se llevó a cabo el estudio de mezclas de Cr(III) y Cr(VI), para examinar la
posible determinación de Cr Total. En la Figura 44, puede observarse el solapamiento
entre las curvas potenciales de Cr(VI) y Mezcla (Cr(VI)+Cr(III)) a dos niveles de
concentración de HCl utilizado en el tratamiento de reducción (HCl 1· 10-3
M y HCl 5· 10-
3
M). Las curvas de calibrado potenciales de las mezclas dieron pendientes similares a las
de las curvas de calibrado potenciales de Cr(VI), cuando se expresa la concentración de
Cromo como Cromo Total (más información puede verse en el Apéndice 7, [77]). De
estos resultados, se deduce que la conversión de Cr(VI) a Cr(III) después del tratamiento
de reducción, es del 100 %.
0
1000
2000
3000
4000
Señalquimioluminiscente
0 g/L
3 g/L
6 g/L
10 g/L
15 g/L
µ
µ
µ
µ
µ](https://image.slidesharecdn.com/meseguer-170216182421/85/Meseguer-136-320.jpg)

![REACCIÓN DEL LUMINOL
116
Tabla 49. Curvas de calibrado simuladas y reales utilizando modelos de calibración
polinómicos.
Conc.
Cr (µµg/l)
Datos simulados Datos experimentales
0 y = 0.266· X2
+ 105.7· X + 2402.2 y = 0.4688· X2
+ 71.158· X - 134.16
5 y = 0.266· X2
+ 108.36· X + 2937.4 y = 0.4076· X2
+ 79.802· X + 182.53
10 y = 0.266· X2
+ 111.02· X + 3485.8 y = 0.3646· X2
+ 86.627· X + 557.03
15 y = 0.266· X2
+ 113.68· X + 4047.5 y = 0.3056· X2
+ 97.317· X + 1423
20 y = 0.266· X2
+ 116.34· X + 4622.6 y = 0.266· X2
+ 105.7· X + 2402.4
En el caso de que hubiese efecto matriz, en los datos polinómicos simulados,
ninguno de los coeficientes del modelo se conservarían, con lo que se descartó para su
evaluación.
Con estos resultados, se concluyó que no se podían utilizar este tipo de curvas
para la evaluación del efecto matriz, siendo únicamente posible diagnosticarlo si se
utilizaban los intervalos lineales de concentración por comparación de las pendientes de
calibración obtenidas.
S Muestra de HCl
El estudio de las rectas de adición estándar en una muestra de HCl que estaba
contaminada con todos los interferentes estudiados, dio porcentajes de recuperación en
pendiente respecto a la curva de calibrado de Cr(VI), entre el 94-114% cuando se trabajó
en condiciones de reducción con HCl 5· 10-3
M y en el intervalo lineal de concentraciones.
Al utilizar el volumen mayor de muestra (AE6) (ver Apéndice 7, [77]), el porcentaje de
recuperación fue del 136%, por estar ya fuera del intervalo lineal (Tabla 50). El valor de
la pendiente de la adición estándar sirve para garantizar que la muestra está dentro del
intervalo lineal de Cromo establecido por el método.
Tabla 50. Pendientes y porcentajes de recuperación para la muestra de HCl.
Adición Estándar b ±± sb %Recuperación
AE1 185 ± 5 93.9%
AE2 200 ± 7 101.5%
AE3 216 ± 3 109.6%
AE4 225 ± 12 114.2%
AE5 193 ± 4 98%
AE6 268 ± 12 136%](https://image.slidesharecdn.com/meseguer-170216182421/85/Meseguer-140-320.jpg)
![Capítulo 2. RESULTADOS OBTENIDOS Y DISCUSIÓN
117
S Muestras de agua
Se analizaron tres muestras de agua y un patrón de referencia certificado.
Las adiciones estándar realizadas a dos volúmenes de una muestra de agua de
grifo en condiciones de reducción con HCl 5· 10-3
M, dieron pendientes similares a las
curvas de calibrado de patrones: (165 ± 7) y (206 ± 4), con porcentajes de recuperación
del 83.7 % y 104.6 % respectivamente, por lo que el MOSA se puede aplicar con
garantía.
La aplicación del método de Youden antes y después del tratamiento de
reducción, dio ordenada en el origen significativa en ambos casos: (170 ± 40) y (100 ±
40). Estos valores habrán de tenerse en cuenta para calcular la concentración de cromo en
las muestras. Se observó que al aplicar el tratamiento de reducción, el valor del Youden
disminuía, con lo que la señal que resultaba de la medida directa, sería debida a Cr(III) y
a otras especies que desaparecían al aplicar el tratamiento de reducción. En los casos en
que se observe este efecto, solo se podrá determinar el Cromo Total.
El estudio de una muestra de agua mineralno proporcionó ninguna señal antes ni
después del tratamiento de reducción, por lo que la muestra no contenía cromo. El
análisis de esta muestra de agua mineral fortificada con 2.5 ìg/l de Cr(III) y 2.5 ìg/l
Cr(VI), por el método quimioluminiscente propuesto y por el método de referencia de la
difenilcarbazida [89], dio resultados similares (Tabla 51). Además los porcentajes de
recuperación del Cromo adicionado a la muestra, fueron entre el 83-92 % utilizando el
método QL, lo que reflejaba la exactitud del método.
Tabla 51. Determinación de Cromo en una muestra de agua mineral.
Método de Referencia
Concentraciones (ìg/l)
Método CL
Concentraciones (ìg/l)
Cr(III) Cr Total Cr(VI) Cr(III) Cr Total Cr(VI)
Muestra (1.5±0.4) (1.7±0.2) - (1.56±0.05) (1.83±0.09) -
Muestra
fortificada
(3.2±0.3) (6.1±0.3) (2.9±0.3) (3.7±0.1) (5.97±0.07) (2.3±0.2)
La validación de la exactitud del método se completó al analizar la muestra de
agua certificada (SRM © 1640), cuyo valor estimado fue (38.2 ± 1) ìg/l de Cromo (III)
frente al valor certificado (38.6 ±1.6) ìg/l Cr(III).](https://image.slidesharecdn.com/meseguer-170216182421/85/Meseguer-141-320.jpg)

![Capítulo 2. RESULTADOS OBTENIDOS Y DISCUSIÓN
119
2.2.1.2. INFLUENCIA DE LOS PROTOCOLOS DE ACONDICIONAMIENTO
DE MUESTRAS DE AGUA EN LA DETECCIÓN QUIMIOLUMINISCENTE DE
ELEMENTOS TRAZA.
La conservación de las muestras es un problema analítico interesante debido a
que la completa estabilidad de todos los constituyentes de la muestra es imposible de
alcanzar. Después de la recogida de la muestra, los procedimientos de conservación
solamente retrasan los cambios químicos y biológicos [89]. Los procesos de adsorción en
la pared de los recipientes ocurren en muestras almacenadas sin acidificar y sin congelar.
Las muestras pueden almacenarse no acidificadas pero congeladas para conservar la
distribución original de las especies [90]. Independientemente de la temperatura de
almacenamiento, muchos autores recomiendan la acidificación inmediatamente después
del muestreo si se quieren analizar iones metálicos.
El análisis inmediato para la determinación de metales en muestras de agua es
siempre recomendable, o bien un tiempo de preservación corto de la muestra filtrada a
baja temperatura (4ºC). En estos casos, la medida se realiza sin ningún otro paso de
acondicionamiento. El análisis de agua embotellada se realiza también sin necesidad de
ningún paso de acondicionamiento, sin embargo, el método más común de
acondicionamiento para la determinación de metales en muestras de agua, es la adición de
un ácido, como el HCl para corregir el pH hasta 2-3. Las muestras previamente filtradas
se almacenan en frascos de plástico a 4ºC, el tiempo máximo recomendado es de 6 meses.
El procedimiento de conservación a largo plazo utiliza la adición de HNO3. Las
muestras se filtran, acidifican y guardan a 4ºC. Las botellas de polietileno se introducen
en bolsas de plástico aluminizado. El tiempo de almacenaje puede durar incluso 2-3 años.
Este es el protocolo de acondicionamiento que siguen los materiales de referencia
certificados.
La quimioluminiscencia proporciona sensibilidad y selectividad y acoplada a la
inyección en flujo (FI), rapidez y reproducibilidad en la inyección de la muestra y
mezclado de los reactivos, como se ha demostrado en el apartado anterior. Estos factores,
junto al bajo coste comparado con la técnicas de AAS e ICP, la simplicidad y la
posibilidad de realizar análisis in situ, hace a la determinación quimioluminiscente
combinada con la inyección en flujo, extremadamente atractiva para la determinación de
elementos traza en muestra de agua medioambientales.
En la Tabla 52, se muestra una selección de las determinaciones
quimioluminiscentes de iones metálicos en muestras de agua utilizando inyección en flujo
descritas en la bibliografía [91-108]. La reacción más utilizada es la de oxidación del
luminol con peróxido de hidrógeno en medio básico catalizada por iones metálicos. Para
cuantificar Fe(II), Fe(III), Mn(II) y Cu(II), se requiere un paso de preconcentración,
normalmente por cromatografía de intercambio iónico. Para complejar Mn(II) y Co(II), se
han utilizado enmascarantes como EDTA, citrato sódico, 8-quinolinol o ácido fosfórico.
En función de estas condiciones, los parámetros analíticos de los diferentes
procedimientos se ven afectados.](https://image.slidesharecdn.com/meseguer-170216182421/85/Meseguer-143-320.jpg)
![REACCIÓN DEL LUMINOL
120
Tabla 52. Estudios de la determinación quimioluminiscente en flujo de iones metálicos en
muestras de agua.
Ref. Iones Reacción Pre-
conc.
Consideraciones del método Muestra de
agua
[91] Mn(II) luminol-H2O2 Si Muestra acidificada AcH-AcNH4
+
pH=5.0.
Conc Mn: 0.05-0.5 µg/L
Marina
[92] Fe(II) luminol-H2O2 Si H3PO4 en patrones/muestras pH=6.
Conc Fe: 10-70 µg/L.
-
[93] Co(II)
Cu(II)
Fe(II)
luminol-H2O2 Si Patrones 0.01mol/L HCl. Fase móvil ácido
oxálico pH=4.2. Conc Co: 0.05-4 µg/L. Cu:
20-200 µg/L. Fe semicuantitativo.
-
[94] Fe(III) sulfoflavina-
H2O2
Si Patrones 5⋅10-3
mol/L HCl. Muestra acidif. a
pH=5.5. Conc Fe: 0.02-5.6 µg/L.
Marina
[95] Fe(III) luminol-H2O2 Si Muestra acidif. AcH-AcNH4
+
a pH=3.0.
Conc Fe(III): 0.005-0.1 µg/L.
Oceánica
[96] Cr(VI) luminol-hexa -
cianoferrato(II)
Si No acondicionamiento.
Conc Cr: 0.04-1 µg/L.
Residual
[97] Co(II) Ácido gálico-
H2O2
Si Muestras en ac fórmico/formato amónico a
pH=3.6. Conc Co: 0.0006-1 µg/L.
Marina de
referencia
[98] Mn(II) luminol-H2O2 Si Patrones en HCl 0.1mol/L.
Conc Mn: 0.002-110 µg/L.
Marina de
referencia
[99] Fe(II)
Fe(III)
luminol-O2. Si Patrones en HCl a pH=3.5.
Conc Fe: 0.001-1 µg/L.
suelo, río,
lluvia, lago
[100] Fe(II)
Fe(III)
luminol-O2. Si Patrones y muestras 0.1mol/L HCl.
Conc Fe: 0.002-0.56 µg/L.
Oceánica y
marina
[101] Fe(II)
Total Fe
Sulfoflavina-
H2O2
Si Patrones en HCl. Muestras en tampón acetato
pH=4.4. Conc Fe: 0.05-11 µg/L.
Lluvia, río
grifo, costa
[102] Co(II)
Fe(II)
Fe(III)
pirogalol-H2O2-
NaOH,
Si Patrones 0.01 mol/L en HCl purificado.
Conc Co: 5-850 µg/L. Fe: -
estuario
[103] Cr(III)
Fe(II)
Co(II)
luminol - H2O2-
haluro
No Con / sin EDTA. Conc Cr: 0.005-5.2 µg/L.
Fe: 0.6-56 µg/L. Co: 0.06-59 µg/L.
-
[104] Co(II)
Mn(II)
luminol -IO4
-
No Muestras H3PO4. Para deter Mn, 8-quinolinol,
y para determ Co y Mn adic citrato. Conc Co:
0.01-12 µg/L. Mn: 0.02-9 µg/L.
fresca y
contaminada
[82] Cr(III) luminol-H2O2 No Patrones y muestras 10-4
M EDTA y 3⋅10-4
M
H3PO4. Disol. H2O2 10-3
M en EDTA.
Conc Cr: 0.01-6 µg/L.
mineral,
potable,
contaminada
[105] Mn(II) TCNQ-O2-OH-
EosinaY
No No acid. Análisis inmediato. Disol. DDAB
vesicular. Conc Mn: 4 – 4000 µg/L.
potable
[106] Fe(II)
Mn(II)
luminol-IO4
-
No No acidif. o -fenantrolin activador determ. Fe.
TEA agente complejante determ. Mn. Conc.
Fe: 0.003-0.1 µg/L. Mn: 0.005-0.1 µg/L.
Suelo,
potable
[86] Cr(III) luminol-H2O2 No H3PO4 en patrones/muestras pH=3.7. EDTA
10-4
M, portador carbonato 0.1 M y bromuro
0.3 M pH=10.8. Conc Cr: 0.5-500 µg/L.
grifo
[107] Cu(II) fenantrolina-
H2O2
No Patrones en HCl a pH=3. Muestras a pH=2.
Conc Cu: 0.006-0.45 µg/L.
marina
[88] Cr(III) luminol-H2O2 No EDTA 10-3
M en patrones/muestras.
Conc Cr: 5.2-5200 µg/L
natural
[108] Cr(III) luminol-H2O2 No Portador EDTA- CO3
-
pH 10.8. Muestras en
condic ácidas. Conc Cr: 0.5-50 µg/L.
río, grifo y
potable](https://image.slidesharecdn.com/meseguer-170216182421/85/Meseguer-144-320.jpg)
![Capítulo 2. RESULTADOS OBTENIDOS Y DISCUSIÓN
121
En la bibliografía no existen referencias que estudien la influencia de las
condiciones de acondicionamiento de las muestras en la señal quimioluminiscente. En
este apartado, se estudia la señal quimioluminiscente directa producida por la reacción del
luminol-peróxido de hidrógeno catalizada por iones metálicos como Cr(III), Co(II),
Fe(II), Fe(III), Ni(II) y Mn(II) en función del protocolo de acondicionamiento de la
muestra. Se han evaluado muestras sin acidificar o acidificadas con HCl o HNO3, tanto en
estático como en flujo, y en presencia o ausencia de EDTA. No se ha utilizado ningún
procedimiento de preconcentración.
El montaje de inyección en flujo utilizado es el que se muestra en la Figura 17
Pág. 40 del Capítulo 1-Introducción. Más información acerca de las condiciones
experimentales se refleja en el Apéndice 8 [78].
Señal QL de muestras no acidificadas (sin acondicionar)
Se estudió inicialmente la señal quimioluminiscente de patrones preparados en
agua ultrapura. La legislación especifica los niveles de concentración máximos
permitidos para los metales en estudio en: 200 µg/L para Fe, 20 µg/L para Ni, 2 mg/L
para Cu y 50 µg/L para Cr [40]. La composición de un agua natural en componentes traza
puede ser descrita por el material de referencia certificado SRM © 1640 (Tabla 53).
Tabla 53. Composición del material de referencia certificado SRM © 1640: agua dulce.
Fracciones de masa certificadas
Fracciones de masa de
referencia
El
em
ent
o
µµg/
Kg
Ele
men
to
µµg/
Kg
Elemento µµg/Kg
Aluminio 52.0 ± 1.5 Cobalto 20.28 ± 0.31 Cobre 34.3 ± 1.6
Antimonio 13.79 ± 0.42 Hierro 34.3 ± 1.6 Litio 27.89 ± 0.14
Arsénico 26.67 ± 0.41 Plomo 27.89 ± 0.14 Níquel 27.4 ± 0.8
Bario 148.0 ± 2.2 Manganeso 121.5 ± 1.1 Potasio 994 ± 27
Berilio 34.94 ± 0.41 Molibdeno 46.75 ± 0.26 Rubidio 2.00 ± 0.02
Boro 301.1 ± 6.1 Selenio 21.96 ± 0.51 Zinc 53.2 ± 1.1
Cadmio 22.79 ± 0.96 Plata 7.62 ± 0.25 Calcio 7.045 ± 0.089
Cromo 38.6 ± 1.6 Estroncio 124.2 ± 0.7 Magnesio 5.819 ± 0.056
Vanadio 12.99± 0.37 Silicio 4.73 ± 0.12
Sodio 29.35 ± 0.31
Talio* < 0.1
* Valor de Fracción de masa informativo.](https://image.slidesharecdn.com/meseguer-170216182421/85/Meseguer-145-320.jpg)
![REACCIÓN DEL LUMINOL
122
Se midió inicialmente la quimioluminiscencia utilizando el sistema de inyección
en flujo. Ni(II) y Mn(II) dieron señales QL similares a las del blanco. Fe(II) y Fe(III)
solo dieron señales significativas a niveles superiores a los límites legislados (0.2 mg/l),
por lo que sería necesario utilizar un paso de preconcentración para aplicar el método a la
determinación de estos metales. Cr(III), Co(II) y Cu(II) dieron señales significativas para
su cuantificación.
Las curvas de calibrado doble-logarítmicas y en los intervalos lineales de
concentración, en las diferentes condiciones experimentales ensayadas, se muestran en la
Tabla 54. Se utilizó la altura de pico como señal analítica. El intervalo lineal de
concentraciones fue: para Cr(III) 3-15 µg/L, para Co 2-8 µg/L, y para Cu, hasta 20 µg/L.
En ausencia de EDTA, los límites de detección calculados para una relación señal/ruido
de 3, estuvieron entre 0.5-2 µg/L. Estos valores son similares a los que se obtienen
utilizando absorción atómica electrotérmica para elementos traza [109].
También se estudió el uso de EDTA como agente complejante de interferentes,
añadiéndolo a la muestra o al portador (tampón o KOH), de forma que el complejo metal-
EDTA se formaba antes o durante el sistema de flujo respectivamente. En presencia de
EDTA, se obtiene que la sensibilidad del Co(II) disminuye drásticamente. Para el Cu(II),
la señal desapareció completamente.
La sensibilidad del Cr(III) disminuyó al adicionar EDTA 0.01 M al portador,
como puede verse en la Tabla 54. Al utilizar EDTA 5· 10-3
M en el portador (tampón), se
observó que la sensibilidad para el Cr(III) era similar a la obtenida en ausencia de EDTA,
por lo que en estas condiciones, la sensibilidad se mantenía, y la reacción era además
selectiva para Cr(III).
Para el procedimiento en estático, las curvas de calibrado para Cromo y Cobalto
en presencia de EDTA 3· 10-3
M, se muestran en la Tabla 55. Como puede verse, la
sensibilidad del Cr resultó 3 veces superior a la del Co. Al adaptar el montaje FIA al
fluorímetro utilizado para las medidas en estático, la curva de calibrado que se obtuvo
para Cr fue: y = (0.07 ± 0.05) + (0.30 ± 0.02)· C (r2
= 0.997, sy/x = 0.07), por lo que la
pérdida de sensibilidad del procedimiento en flujo respecto al procedimiento en estático,
fue del orden del 60%.](https://image.slidesharecdn.com/meseguer-170216182421/85/Meseguer-146-320.jpg)




![Capítulo 2. RESULTADOS OBTENIDOS Y DISCUSIÓN
127
Aplicabilidad
La señal quimioluminiscente variaba en función del protocolo de
acondicionamiento de la muestra y de la presencia de agente enmascarante, aunque esta
influencia era mínima para el Cr(III). Añadiendo EDTA se obtuvo especificidad para la
determinación de Cromo, sin que se requiriera neutralización al utilizar el procedimiento
en flujo. Según las condiciones experimentales seleccionadas, la reacción resultó
selectiva para la determinación de Cr(III), o bien se puede evaluar la contribución total de
Cr, Co y Cu. En la Figura 47, se muestran las pendientes obtenidas para cada uno de los
metales estudiados en las diferentes condiciones ensayadas utilizando el procedimiento en
flujo. El método en estático requirió la adición de Na2CO3 al acondicionar las muestras en
condiciones ácidas drásticas (HNO3 0.5M).
Figura 47. Pendiente de la curva de calibrado en el intervalo lineal en las diferentes
condiciones experimentales. 1: Sin acondicionar; 2: HCl 5· 10-3
; 3: HNO3 0.5; 4: EDTA 10-2
; 5:
EDTA 1· 10-2
, HCl 5· 10-3
; 6: EDTA 5· 10-3
; 7: EDTA 5· 10-3
, HCl 5· 10-3
; 8: EDTA 10-2
(portador
KOH); 9: EDTA 10-2
(portador tampón); 10: EDTA 5· 10-3
(portador KOH); 11: EDTA 5· 10-3
(portador tampón); 12: EDTA 10-2
(portador tampón), HCl 5· 10-3
; 13: EDTA 5· 10-3
(portador
KOH), HCl 5· 10-3
; 14: EDTA 5· 10-3
(portador tampón), HCl 5· 10-3
. Concentración: mol/L.
Para estudiar la señal quimioluminiscente total, se llevaron a cabo varias
experiencias, con la intención de evaluar la aditividad de las señales en las diferentes
condiciones estudiadas. Se estudiaron mezclas binarias y terciarias de los tres metales
almacenados en condiciones ácidas y no ácidas (ver parte experimental en el Apéndice 8,
[78]). Las mezclas dieron porcentajes de recuperación de señal próximos al 100%
(Figura 48), demostrándose así la aditividad entre las señales de los tres metales tanto en
estático (mezclas s1, s2, s3, s4) como en flujo (mezclas s5, s6, s7, s8, s9, s10, s11, s12,
s13, s14, s15, s16).
0
100
200
300
400
500
600
700
800
1
2
3
4
5
6
7
8
9
10
11
12
13
14
Acondicionamiento de la muestra
b(L/mg)
Cr
Co
Cu](https://image.slidesharecdn.com/meseguer-170216182421/85/Meseguer-151-320.jpg)


![REACCIÓN DEL LUMINOL
130
2.2.1.3. CALIBRACIÓN MULTIVARIADA APLICADA A LA DETERMINACIÓN
QUIMIOLUMINISCENTE SIMULTÁNEA DE COBALTO Y CROMO.
En la bibliografía se encuentran numerosos métodos analíticos propuestos para la
determinación de cromo y/o cobalto, como la electroforesis capilar [110], NAA [111],
XRF [112-113], AAS[114-115], ICP-AES [116], o ICP-MS [112,117].
Se han propuesto diferentes estrategias para la determinación selectiva o
simultánea de iones metálicos utilizando la reacción quimioluminiscente del luminol. En
los sistemas de inyección en flujo, se han utilizado pre-tratamientos o procedimientos de
separación. Se ha publicado el uso de agentes enmascarantes [104], separadores de
membrana [118] o resinas de intercambio iónico [119]. Para la determinación simultánea
de varios metales se ha propuesto la cromatografía iónica con detección
quimioluminiscente [120,81]. Sin embargo, en este caso, la limitación es la
incompatibilidad de la fase móvil con las condiciones de reacción requeridas para la
reacción postcolumna [93].
Se han resuelto mezclas de compuestos orgánicos [121-122] y de iones metálicos
[123-124] por calibración multivariada y/o la técnica de eliminación de interferentes. El
método de mínimos cuadrados parciales (PLS) se ha aplicado a la determinación
simultánea de mezclas de compuestos en muestras de agua.
En este apartado, se estudia un modelo de mínimos cuadrados parciales (PLS)
para la determinación simultánea de Cr(III) y Co(II) en muestras de agua, registrando en
estático la curva cinética, y construyendo diferentes modelos a partir del conjunto de
medidas señal-tiempo. La capacidad de predicción se ha evaluado utilizando disoluciones
patrón, y el modelo ha sido validado en la determinación de concentraciones de Cr(III) y
Co(II) en un material de referencia certificado de agua natural.
Más información acerca de las condiciones experimentales, se refleja en el
Apéndice 9 [125].
Estudio de la señal quimioluminiscente
Cuando se trabaja en estático, la emisión quimioluminiscente del sistema
luminol/H2O2 describe una curva de respuesta Intensidad frente al Tiempo de primer
orden [126]: formación y destrucción del emisor de luz (3-aminoftalato en este caso). La
velocidad de reacción aumenta con la concentración de diferentes iones metálicos.
La curva de respuesta depende de factores experimentales como el pH o la
velocidad de mezclado [127], pero su influencia se evitó manteniéndolos constantes.
También depende del metal presente en la disolución, siendo Cr y Co los metales que
contribuyen principalmente a la señal quimioluminiscente en las condiciones estudiadas,
tal como se demuestra en el capítulo anterior. La contribución de otras especies se
consideró despreciable.](https://image.slidesharecdn.com/meseguer-170216182421/85/Meseguer-154-320.jpg)





![Capítulo 2. RESULTADOS OBTENIDOS Y DISCUSIÓN
137
Las novedades que aporta esta publicación frente a otras publicaciones existentes
consisten en que:
§ Se ha demostrado la robustez de los perfiles intensidad-tiempo.
§ Es la primera aplicación de estos perfiles intensidad-tiempo en calibración
multivariada para el análisis quimioluminiscente simultáneo de dos especies. Otras
publicaciones utilizan registros intensidad-longitud de onda obtenidos a partir de
dispositivos especiales (DDC) para determinar un analito [128-129].
§ Se han validado los resultados con un material de referencia certificado.](https://image.slidesharecdn.com/meseguer-170216182421/85/Meseguer-161-320.jpg)
![REACCIÓN DEL LUMINOL
138
1.2. DETERMINACIÓN DE NITRÓGENO ORGÁNICO AMINO Y
AMONIACAL.
El Nitrógeno es un nutriente clave en aguas naturales, pero los vertidos de
nitrógeno en exceso puede causar procesos de eutrofización de las aguas. Las especies
dominantes de nitrógeno en las aguas son: el nitrógeno inorgánico disuelto (amonio,
nitrito y nitrato), el nitrógeno orgánico disuelto (la fracción más importante está
constituida por aminoácidos y péptidos, y se le denomina habitualmente N-amino), el
Nitrógeno particulado orgánico (debido a pequeños organismos: algas, bacterias) y el
particulado inorgánico [130]. La presencia de Nitrógeno Orgánico Disuelto (DON) en
aguas naturales es en gran medida debido a procesos biológicos autóctonos.
Adicionalmente, las fuentes externas de DON provienen de aguas residuales industriales,
migraciones terrestres o deposición atmosférica [131]. La concentración de DON en
aguas se ha ignorado durante mucho tiempo. Su medida resulta dificultosa y su carácter
químico se ha descrito escasamente. Esto, unido a que se supone biológicamente inerte,
pueden ser las causas [131].
Existen buenos procedimientos analíticos para cuantificar el nitrógeno inorgánico
disuelto (DIN) y el nitrógeno disuelto total (TDN), pero no para el Nitrógeno orgánico
disuelto, tal como se ha mencionado anteriormente. El más utilizado es el método
Kjeldahl [132-135], debido a que es el de referencia establecido por la Directiva Europea
[40] relativa a los métodos de medida en el análisis de aguas superficiales. El método
Kjeldahl se desarrolló específicamente para el nitrógeno proteico, y algunos autores han
indicado la recuperación incompleta del nitrógeno para algunos compuestos [136-137].
Este método proporciona el contenido de Nitrógeno orgánico disuelto y amonio.
En otros protocolos el DON se calcula como la diferencia entre las medidas
independientes del TDN y el DIN, pero estos métodos son incluso más largos en tiempo
que el método de Kjeldahl. Los principales métodos para la determinación del TDN son:
la oxidación alcalina con persulfato o la fotooxidación-UV a nitrato y los métodos de
combustión oxidativa a NO [131,136].
Se han publicado algunos trabajos comparando varios de estos métodos [131,138-
139], pero se necesita todavía mucho más trabajo para mejorar la exactitud de los
resultados de los métodos para el DON y reducir el tiempo y/o la dificultad de la técnica.
En esta parte de la Tesis, se ha profundizado en el estudio del Nitrógeno Orgánico
en dos direcciones:
§ En una primera parte, se han demostrado las ventajas del método fluorimétrico de
OPA/NAC frente al Nessler para la determinación de amonio después de la digestión
Kjeldahl, además de demostrar que puede estimar directamente amonio y aminas
primarias.
§ En la segunda parte, se propone un método quimioluminiscente que puede evaluar el
nitrógeno orgánico mayoritario total de forma rápida y automática.](https://image.slidesharecdn.com/meseguer-170216182421/85/Meseguer-162-320.jpg)
![Capítulo 2. RESULTADOS OBTENIDOS Y DISCUSIÓN
139
2.2.2.1. MÉTODO FLUORIMÉTRICO DE ROTH MODIFICADO PARA
MUESTRAS DE AGUA: DETERMINACIÓN SIMULTÁNEA DE AMONIO Y
GRUPOS AMINO PRIMARIO Y SU UTILIDAD EN LA DETERMINACIÓN DEL
NITRÓGENO KJELDAHL TOTAL.
El amonio es un micronutriente en los sistemas acuosos, y tiene un papel
fundamental en el ciclo del nitrógeno de los ecosistemas acuáticos. Sin embargo, su
determinación todavía es una tarea delicada, debido a su susceptibilidad a la
contaminación, y a que los métodos clásicos no parecen estar bien controlados en la
mayoría de los laboratorios [140]. Habitualmente, el amonio se encuentra junto a aminas
alifáticas de cadena corta (principalmente metilamina), debido a que estas se hallan
ampliamente distribuidas en el medioambiente. Los límites tolerables para aminas se
regulan como el Nitrógeno Kjeldahl Total (que incluye amonio y Nitrógeno Orgánico),
siendo los valores habituales entre 15 y 85 mg/L para aguas residuales, y 1 mg/L para
agua de grifo.
La cromatografía líquida combinada [10] o no con derivatización [141], se utiliza
generalmente para el análisis de amonio y aminas alifáticas en medio acuoso. En la
bibliografía no se encuentran métodos UV-VIS o fluorimétricos libres de errores
sistemáticos que puedan utilizarse como métodos de screening de muestras para reducir
costes y tiempo de análisis en el laboratorio medioambiental, o para determinaciones in
situ. El método fluorimétrico de Roth [142-144] para la determinación de aminas
primarias utilizando como reactivos derivatizantes un tiol y el o-ftaldialdehido (OPA),
podría adaptarse a la determinación de amonio y grupos amino primario totales.
Aprovechando la respuesta selectiva de amonio con el reactivo OPA-NAC a 415 nm, que
se ha demostrado en el Punto 2.1.2.1 del Capítulo 2-Resultados de esta Tesis (Apéndice
4, [55]), y que nuestro grupo de investigación ha publicado recientemente la
determinación de grupos amino primario en muestras de agua llevando a cabo la
derivatización de las aminas en cartuchos C18 y utilizando el reactivo OPA-NAC
[145,37] y 333 nm como longitud de onda de excitación, se estudió en este trabajo la
determinación simultánea de amonio y metilamina, como compuesto modelo de los
grupos amino primario, utilizando el reactivo OPA-NAC como agente derivatizante,
excitando a dos longitudes de onda 415 nm y 333 nm, y empleando una metodología de
calibración tanto bivariada como multivarida (regresión en componentes principales).
El método se aplicó a patrones antes y después del tratamiento Kjeldahl, siendo
un buen indicador del nitrógeno amino primario presente en la muestra o un buen método
para amonio después del tratamiento Kjeldahl, y también un buen indicador de fallos en
el tratamiento de digestión Kjeldahl utilizado para la conversión a amonio de los grupos
amino primario. El método se aplicó al análisis de muestras de agua reales, y los
resultados se compararon con los obtenidos al aplicar el método de referencia de Nessler.
Más información acerca de las condiciones experimentales, se refleja en el
Apéndice 10.](https://image.slidesharecdn.com/meseguer-170216182421/85/Meseguer-163-320.jpg)
![REACCIÓN DEL LUMINOL
140
Etapa de calibración
De acuerdo con el trabajo previo del grupo de investigación [145], los patrones de
MA, EA, N-PrA, IPA, BA, PeA, HA y β-FEA, proporcionaron respuestas similares entre
ellos al expresar la concentración en mg/L de Nitrógeno, con porcentajes de recuperación
respecto a la señal de la MA de 100, 93.5, 101.1, 93.4, 89, 100.4, 108.2 y 99 %
respectivamente. La aditividad entre la señal de diferentes aminas se evaluó analizando
mezclas de estas. Los porcentajes de recuperación fueron entre el 81-88 %. Con estos
resultados se demostró que la respuesta de las aminas en la reacción con OPA-NAC, no
dependía de la amina primaria, y se podía utilizar la MA como compuesto modelo.
Para estudiar la determinación simultánea de amonio y aminas primarias (MA),
se empleó el diseño experimental 52
que se muestra en la Tabla 63. Tanto el amonio
como la amina primaria proporcionaban una señal fluorescente a ëexc = 333 nm / ëem =
450 nm, aunque el máximo de emisión para el amonio se situaba a 462 nm (Figura 53).
Tabla 63. Código y concentración del conjunto de patrones de calibración (diseño 5
2
).
NH4
+
MA
(mg/L)
0 0.25 0.375 0.5 0.625
0 00 01 02 03 04
0.25 10 11 12 13 14
0.75 20 21 22 23 24
1.25 30 31 32 33 34
1.75 40 41 42 43 44
No hubo diferencias significativas a un nivel de confianza del 95% (varianza
homogénea, Fcalc < Ftab, α=0.05 , y pendientes comparables, tcalc < ttab, α=0.05 ) entre las curvas
de calibrado de amina obtenidas en presencia de diferentes concentraciones de amonio
constante variado de 0 a 1.75 mg/L (Tabla 64). Las pendientes medias obtenidas a 450
nm para amina y amonio fueron 6532 (s= 472, n= 5) y 1038 (s= 127, n= 5), y expresando
la concentración en mg/L de N, 2950 (s= 213, n= 5) y 855 (s= 104, n=5),
respectivamente. Estos valores indican que el isoindol de amina es 3.5 veces más sensible
que el isoindol de amonio cuando se trabaja excitando a 333 nm y midiendo la emisión a
450 nm.](https://image.slidesharecdn.com/meseguer-170216182421/85/Meseguer-164-320.jpg)


![Capítulo 2. RESULTADOS OBTENIDOS Y DISCUSIÓN
143
Los espectros de emisión obtenidos excitando a 415 y 333 nm para los patrones
(Tabla 63), se procesaron utilizando regresión en componentes principales (PCR) para
desarrollar un modelo multivariado para amonio y amina respectivamente. Para llevar a
cabo la etapa de calibración, se emplearon la Selección Top Down (TDS) y la selección
del mejor Sub-Conjunto (BSS) [146]. Los modelos de PCR se obtuvieron utilizando los
datos (centrados) como bloque-X. El número de variables, el porcentaje de varianza
explicada, el número de componentes y la media cuadrática de los errores de predicción
(RMSECV) por validación cruzada, se muestran en la Tabla 65. Los valores de
RMSECV se calcularon para evaluar la capacidad de predicción de los modelos. Para la
metilamina no había diferencias entre las dos opciones de selección, y se obtuvo un
modelo de complejidad 3. Para el amonio, la componente principal 2, no se incluyó en el
modelo con selección BSS, utilizándose solamente las componentes principales 1 y 3
(complejidad 2). En un segundo paso, se llevó a cabo una selección de las variables sobre
el modelo optimizado utilizando la aproximación de Eliminación de Variables No
informativas (UVE) [147]. Se utilizaron solamente 32 y 29 de las variables originales
para metilamina y amonio respectivamente. En ambos casos, se seleccionó un modelo
final de complejidad dos. Los valores de RMSECV fueron buenos para todos los
modelos, como puede verse en la Tabla 65, pero se seleccionaron los modelos de UVE-
BSS.
Tabla 65. Características de los modelos de Regresión en Componentes Principales
obtenidos para amonio y metilamina utilizando Selección Top Down (TDS), Selección del
mejor Subconjunto (BSS) o Eliminación de las Variables no informativas-Selección del mejor
Subconjunto.
Amonio (ëexc 415 nm) Amina (ëexc 333 nm)
TDS BSS UVE-BSS TDS BSS UVE-BSS
Número de
variables
231 231 32 180 180 29
CPs 1,2 1,3 1,2 1,2,3 1,2,3 1,2
Varianza
Explicada,
bloque-X
99.999 99.999 99.999 99.999 99.999 99.999
RMSECV 0.089 0.051 0.052 0.034 0.037 0.040
Precisión y exactitud
Teniendo en cuenta los resultados previos, se analizaron mezclas de patrones de
amonio y metilamina. Las medidas se llevaron a cabo excitando a 415 nm y a 333 nm, y
utilizando las curvas de calibrado de amonio a 485 nm y las curvas de calibrado de
amonio y de metilamina a 440 nm. En la Tabla 66 se muestran las concentraciones de
amonio y metilamina calculados en las diferentes mezclas ensayadas para cada uno de los
modelos de calibración utilizados. Los resultados obtenidos utilizando calibración
bivariada fueron satisfactorios en todos los casos, siendo los errores relativos entre 4-24%](https://image.slidesharecdn.com/meseguer-170216182421/85/Meseguer-167-320.jpg)






![REACCIÓN DEL LUMINOL
150
2.2.2.2. MÉTODO QUIMIOLUMINISCENTE AUTOMÁTICO PARA LA
ESTIMACIÓN TOTAL DEL NITRÓGENO ORGÁNICO (-CH2-NH-) Y AMONIO.
Basándonos en la bibliografía reciente para amonio [148] y en la conocida
reactividad entre los grupos amino y el hipoclorito, en este capítulo se propone un sistema
de flujo continuo para la determinación conjunta del nitrógeno orgánico amino (-CH2-
NH-) y el nitrógeno amoniacal en muestras de agua no tratadas. En este sistema, el grupo
amino reacciona con el Hipoclorito formando la cloramina, y posteriormente, la mezcla
reacciona con el Luminol generando una señal quimioluminiscente. La señal de emisión,
registrada a 425 nm en función del tiempo, disminuye al aumentar la concentración de
Nitrógeno en la muestra, debido a la disminución en la concentración de Hipoclorito, que
depende de la concentración de grupos amino presentes en la muestra.
Se han ensayado un gran número de compuestos de Nitrógeno, y sus
sensibilidades se han comparado en (mg/L)-1
de Nitrógeno. Una misma curva de
calibrado puede utilizarse para un gran número de compuestos, con lo que el método
puede emplearse para deducir de forma rápida el Nitrógeno orgánico disuelto y el
amonio. Se han evaluado las características del método como la selectividad, tiempo de
análisis y reproducibilidad.
El método quimioluminiscente se ha aplicado al análisis de una amplia variedad
de muestras de agua (agua natural, de lago, de riego, de fuente, residual y marina). La
exactitud del método se ha evaluado aplicando el método de adición estándar y
procedimientos de referencia. Para algunas de estas muestras, se ha realizado un estudio
comparativo del contenido de Nitrógeno amoniacal antes y después del tratamiento
Kjeldahl, para evaluar si el método aplicado a muestras no tratadas podía dar una
estimación del N Kjeldahl Total.
El sistema de inyección en flujo utilizado es el que se muestra en la Figura 18 del
Capítulo 1-Introducción (Pág. 41). Más información acerca de las condiciones
experimentales, se refleja en el Apéndice 11.
Parámetros analíticos
En la Tabla 70, se muestran las curvas de calibrado Señal QL frente a
Concentración de Nitrógeno (mg/L) obtenidas con patrones de amonio en diferentes
condiciones experimentales. Las variables evaluadas fueron: el tipo de celda de medida
(celda de nudos o espiral), la concentración de tampón, la concentración de hipoclorito, y
la presencia de tampón o de NaCl en los patrones.
Las condiciones óptimas, de máxima sensibilidad (marcadas en negrita en la
Tabla 70), se obtuvieron utilizando la celda de nudos e Hipoclorito 0.9 mM en tampón
carbonato 0.144 mM de pH 11.2. El rango lineal fue de 0.23-4 mg/L N, y el límite de
detección alcanzado fue de 0.070 mg/L N. Al acondicionar los patrones con tampón, la
sensibilidad disminuyó un 33%, y al prepararlos en NaCl, disminuyó un 13%.](https://image.slidesharecdn.com/meseguer-170216182421/85/Meseguer-174-320.jpg)




![Capítulo 2. RESULTADOS OBTENIDOS Y DISCUSIÓN
155
El aminoácido Histidina presenta una estructura con dos nitrógenos aromáticos
además del grupo amino alifático, y al igual que para la triazina, la señal
quimioluminiscente del blanco aumentó y no se observó disminución de señal en el
intervalo de trabajo del Nitrógeno. Por su parte, el aminoácido Arginina que presenta
Nitrógenos resonantes además del grupo amino alifático, los resultados obtenidos
considerando solamente el nitrógeno alifático (baminoácido*100/bamonio=96.8%), confirmaron
que solo este grupo (–CH2-NH2) de la arginina contribuyó a la disminución de la señal
quimioluminiscente. Los aminoácidos Cisteína y Metionina dieron porcentajes de
recuperación (baminoácido*100/bamonio) de 287 y 147 %. Estos porcentajes indicaron que otro
átomo de su estructura contribuía a disminuir la señal quimioluminiscente. Como se
demuestra más abajo, este átomo fue el S.
Selectividad del método: Estudio de interferencias
El tipo de compuestos interferentes estudiados fueron aquellos que contenían
átomos de sulfuro, ya que estos átomos podían dar lugar a interferencias en la
determinación de nitrógeno como se había visto en la sección anterior al estudiar los
aminoácidos que contenían azufre en su estructura. Se estudió la respuesta de dos
compuestos, el sulfuro sódico (S2-
) y del ácido mercaptoacético (R-SH) que no contenían
grupos nitrógeno en su estructura. Ambos compuestos respondieron a la reacción
disminuyendo la señal QL. La interferencia del sulfuro fue el doble que la del
mercaptoacético, siendo las pendientes de las curvas de calibrado 3058 y 1850 L/mg S.
La respuesta de aminoácidos como la Cisteína (aminoácido con grupo R-SH en
su estructura) y su derivado la N-Acetil-L-Cisteína, era debida a la contribución de ambos
átomos, N y S. Las señales experimentales fueron próximas al 100% de las señales
teóricas obtenidas teniendo en cuenta las pendientes de calibración de amonio y
mercaptoacético. La interferencia causada por el grupo R-S-R, es pequeña, como puede
deducirse del porcentaje de recuperación dado en la sección anterior para la metionina.
Cuando se trabajó a la concentración habitual de tioles en aguas (0.25 µg/L) [149], y a la
concentración máxima permitida para H2S en aguas de bebida (50 µg/L) [40], los
compuestos de azufre no interfirieron en la reacción, obteniéndose una señal
quimioluminiscente igual a la del blanco.
El segundo grupo de posibles interferentes ensayado fueron los iones metálicos.
Cr(III) y Co(II) no interfirieron en la reacción incluso a niveles de 0.5 mg/L. Cu(II)
solamente disminuyó la señal QL en un 10% cuando se utilizó a una concentración de 50
µg/L (límite máximo permitido para aguas naturales). Otros cationes como Fe(III), Cd(II)
y Hg(II), no interfirieron a su máxima concentración permitida (0.5 mg/L, 5 µg/L y 1µg/L
respectivamente).](https://image.slidesharecdn.com/meseguer-170216182421/85/Meseguer-179-320.jpg)


![REACCIÓN DEL LUMINOL
158
Además, se determinó el contenido de Nitrógeno de algunas de estas muestras de
agua después de aplicar el tratamiento Kjeldahl. Los resultados de la Tabla 76 muestran
la presencia de dos grupos de muestras:
- Por una parte, las muestras cuyo contenido de Nitrógeno es el mismo antes y después
del tratamiento Kjeldahl (como es el caso de las muestras S6 y S7). La muestra S6 se
analizó además por los métodos de Nessler y o-ftaldialdehido/N-Acetil-L-Cisteína
(OPA/NAC) para amonio que se desarrolla en el Punto 2.1.2.1 del Capítulo 2-
Resultados de esta Tesis (Apéndice 4, [55]), y después del tratamiento Kjeldahl, las
concentraciones halladas fueron: (180 ± 3, n=3) y (166 ± 3, n=3) mg/L N para los
métodos de Nessler y OPA/NAC respectivamente. En este tipo de muestras, el
tratamiento de Kjeldahl podría haberse sustituido por la aplicación sencilla y directa
delsistema FIA presentado en este trabajo.
- Por otra parte, las muestras que presentaban concentración de Nitrógeno diferente
antes y después del Kjeldahl (como es el caso de las muestras S4, S5 y S8). La
muestra S4 presentó de forma directa el 60% de la señal del N-Kjeldahl; la muestra
S5 no podía analizarse de forma directa porque presentaba un aumento de señal
respecto al blanco como se ha expuesto previamente; y en cuanto a los resultados
para la muestra S8, las diferencias podrían explicarse debido a la presencia de
compuestos de Nitrógeno que no reaccionaban con el hipoclorito pero que se
transformaban en amonio después del tratamiento Kjeldahl, o más probablemente
debido a la presencia de matrices orgánicas complejas que se descomponían en la
digestión ácida (de forma similar a los resultados de la albúmina). Esta muestra, S8,
se analizó además por los métodos de Nessler y OPA-NAC para amonio (Apéndice 4,
[55]), siendo los resultados (0.25±0.02, n=3) y (0.29±0.02, n=3) mg/L N,
respectivamente antes del tratamiento Kjeldahl, y (1.66 ± 0.02, n=3) y (1.635 ± 0.18,
n=3) mg/L N, respectivamente después del tratamiento Kjeldahl. Estos resultados son
bastante similares a los obtenidos con el procedimiento propuesto (Tabla 76).
Conclusiones
La respuesta de los compuestos de Nitrógeno tipo amino (-CH2-NH-) incluido el
amonio, al aplicar la metodología propuesta dio porcentajes de recuperación cercanos al
100% para aminas alifáticas primarias y secundarias, poliaminas, arilaquilaminas, y para
14 de los aminoácidos ensayados. Las aminas terciarias y los Nitrógenos resonantes o los
aromáticos no dieron la reacción, y las proteínas nitrogenadas dieron recuperaciones
incompletas. Los principales interferentes de la reacción fueron los grupos tiol, pero se
demostró que no interferían a la concentración máxima permitida por la legislación. De
los iones metálicos estudiados, ninguno de ellos presentó interferencia a los máximos
niveles permitidos. El método permite hacer una estimación del N-amino Total disuelto
en la muestras de forma muy rápida.](https://image.slidesharecdn.com/meseguer-170216182421/85/Meseguer-182-320.jpg)


![Capítulo 2. RESULTADOS OBTENIDOS Y DISCUSIÓN
161
2.3. REFERENCIAS BIBLIOGRÁFICAS
[1] L.Pan, J.Michael Chong, J.Pawliszyn, J.Chromatogr.A, 773 (1997) 249.
[2] M.Abalos, J.M.Bayona, F.Ventura, Anal.Chem., 71 (16) (1999) 3531.
[3] C.Maris, A.Laplanche, J.Morvan, M.Bloquel, Water.Sci.Technol., 40(6) (1999) 141.
[4] C.Maris, A.Laplanche, J.Morvan, M.Bloquel, J.Chromatogr.A., 846 (1-2) (1999) 331.
[5] F.Sacher, S.Lenz, H.J. Brauch, J.Chromatogr. A., 764 (1) (1997) 85.
[6] H.Wang, J.Lin, X.Liu, H.S.Zhang, Anal.Chim.Acta, 423 (2000) 77.
[7] J.Verdu Andrés, P.Campíns Falcó, R.Herraez Hernandez, Analyst, 126 (2001) 1683.
[8] J.M.You, W.J.Lao, G.J.Wang, Analyst, 124 (12) (1999) 1755.
[9] M. Cobo, M.Silva, J.Chromatogr. A, 848 (1999) 105.
[10] D.Sahasrabuddhey, A.Jain, K.K.Verma, Analyst, 124 (1999) 1017.
[11] M.Yamaguchi, S.Hara, J.Aoki, K.Yoshikuni, T.Iwata, Anal.Sci., 14(2) (1998) 425.
[12] D.Djozan, M.A. Faraj-Zadeh, J. High. Resolut. Chromatogr., 19 (11) (1996) 633.
[13] J.Verdu Andrés, P.Campíns Falcó, R.Herraez Hernandez, Chromatographia, 55
(2002) 129.
[14] W.H.Matchett, W.C.Brumley, J. Liq. Chromatogr.Rel.Technol., 20 (1997) 79.
[15] N.Seiler, Handbook of derivatives for Chromatography, Jonh Wiley, New York,
1998.
[16] J.M Roselfeld., J. Chromatogr. A, 843 (1999) 19.
[17] C.Molins-Legua, P.Campins Falcó, A.Sevillano Cabeza, M. Pedrón Pons, Analyst,
124 (1999) 477.
[18] C.Molins Legua, P.Campíns Falcó, A.Sevillano Cabeza, Analyst, 123(12) (1998)
2871.
[19] R.Herraez Hernandez, P.Campins Falcó, A.Sevillano Cabeza, Anal.Chem., 68(5)
(1996) 734.
[20] D.Shangguan, Y.Zhao, H.Han, R.Zhao, G.Liu, Anal. Chem., 73 (2001) 2054.
[21] S. Meseguer-Lloret, C. Molins-Legua, P. Campins-Falcó, J. of Chromatogr. A, 978
(2002) 59.
[22] C.Molins Legua, P.Campíns Falcó, S. Meseguer Lloret, Chromatographia, 58
(2003) 15.
[23] C.Dodeigne, L.Thunus, R.Lejeune, Talanta, 51 (2000) 415.
[24] T.Kawasaki, M.Maeda, A.Tsuji, J.Chromatogr., 328 (1985) 121.
[25] K.Nakashima, M.Maeda, A.Tsuji, J.Chromatogr., 530 (1990) 154.
[26] S.R.Spurlin, M.M.Cooper, Anal. Lett., 19 (1986) 2277.
[27] J.Ishida, N.Horike, M. Yamaguchi, Anal.Chim.Acta, 302 (1995) 61.
[28] H.Yoshida, R.Nakao, T.Matsuo, H.Nohta, M.Yamaguchi, J. Chromatogr. A. 907
(2001) 39.
[29] J.B.Noffsinger, N.D.Danielson, Anal. Chem., 59 (1987) 865.
[30] X.Wang, D.R. Bobbitt, Talanta, 53 (2000) 337.
[31] Andrew W. Knight, Trends in Anal.Chem., 18 (1999) 47.
[32] M.Cobo, M.Silva, J.Chromatogr. A, 848 (1999) 105.
[33] C.Molins Legua, P.Campins Falcó, A.Sevillano Cabeza, Anal.Chim.Acta, 378 (1999)
83.
[34] P.J.M.Kwakman, H. Koelewijn, I.Kool, U.A.T. Brinkman, G.J.De Jong,
J.Chromatogr,. 511 (1990) 155.](https://image.slidesharecdn.com/meseguer-170216182421/85/Meseguer-185-320.jpg)
![REFERENCIAS BIBLIOGRÁFICAS
162
[35] M.Tod, J.Y.Legendre, J.Chalom, H.Kouwtli, M-Poulou, R.Farinotti, G.Mahuzier,
J.Chromatogr., 594 (1992) 386.
[36] S. Meseguer-Lloret, C.Molins-Legua, J.Verdú-Andrés, P.Campíns-Falcó, J. of
Chromatogr. A, in press.
[37] Y. Moliner-Martínez, C. Molins-Legua, P. Campíns-Falcó, Talanta, 62 (2004) 373.
[38] R. Weinberger, J. of Chromatogr., 314 (1984) 155.
[39] N. Hanoaka, J. of Chromatogr., 503 (1990) 155.
[40] Directiva Europea 98/83/CEE.
[41] Díaz-Santos S.A. Métodos Normalizados para el análisis de agues potables y
residuales. 17 Ed.1992. Apha-Awwe-WPCF
[42] H.Hara, A.Motoike, S.Okazaki, Anal.Chem., 59(15) (1987) 1995.
[43] H.Hara, A.Motoike, S.Okazaki, Analyst, 113(1) (1988) 113.
[44] F.Cañete, A.Ríos, M.D.Luque de Castro, M.Valcárcel, Analyst, 113 (1988) 739.
[45] M.Menezes-Santos, B.Freire-dos-Reis, H.Bergamin, N.Baccan, Anal.Chim.Acta.,
261(1-2) (1992) 339.
[46] H.Shen, T.J.Cardwell, R.W.Cattrall, Analyst, 123 (1998) 2181.
[47] Z.Genfa, T.Uehara, P.K.Dasgupta, A.D.Clarke, W.Winiwarter, Anal. Chem., 70
(1998) 3656.
[48] Y.Huang, Shi-fen Mou, J.M.Riviello, J. of Chromatogr. A., 868(2) (1999) 209.
[49] J.Li, P.K.Dasgupta, Z.Genfa, Talanta, 50 (1999) 617.
[50] W.Qin, Z.Zhang, B.Li, Y.Peng, Talanta, 48 (1999) 225.
[51] J.P.Hart, A.K.Abass, D.C.Cowell, A.Chappel, Electroanalysis, 11(6) (1999) 406.
[52] A. Walcarius, V.Vromman, J.Bessiere, Sens-Actuators-B, B56(1-2) (1999) 136.
[53] H.Mana, V.Spohn, Fres. J. Anal. Chem., 366 (2000) 825.
[54] Schwarz, H. Kaden, G.Pausch, Fres. J. Anal. Chem., 367 (2000) 396.
[55] S. Meseguer Lloret, C. Molins Legua, P. Campins Falcó, Intern. J. of Environ. Anal.
Chem., 82 (2002) 475.
[56] W.J. Youden, Anal. Chem.,19 (1947) 946.
[57] P.Campins-Falco, J. Verdú-Andrés, F. Bosch-Reig, C.Molins-Legua, Anal. Chim.
Acta, 302 (1995) 323.
[58] Instruction manual for ammonia electrode (NH3) (Mettler Toledo GmbH, Process,
Urdorf).
[59] A. Aminot, R.Kerouel, D.Birot, Wat.Res. 35-7 (2001) 1777.
[60] J.P. Hart, A.K. Abass, D.C. Cowell, A. Chappel, Electroanalysis, 11(1999) 406.
[61] J. Schwarz, H. Kaden, G. Pausch, Fres. J. Anal. Chem., 367 (2000) 396.
[62] M-Mori, K.Tanaka, M.I.H.Helaleh, Q.Xu, M.Ikedo, Y.Ogura, S.Sato, W.Hu,
K.Hasebe, J.Chromatogr. A, 997 (2003) 191.
[63] D.H.Thomas, M.Rey, P.E.Jackson, J.Chromatogr. A, 956 (2003) 181.
[64] H.Mana, V.Spohn, Fres. J. Anal. Chem., 366 (2000) 825.
[65] J.Li, P.K.Dasgupta, Anal.Chim.Acta, 398 (1) (1999) 33.
[66] W.Qin, Z.J.Zhang, B.X.Li, Y.Y.Peng, Talanta, 48 (1999) 225.
[67] M.Marcé, D.S.Brown, T.Capell, X.Figueras, A.F.Tiburcio, J. of Chromatogr. B:
Biomed. Sci. and Appl., 666 (2) (1995) 329.
[68] M.Cobo, M.Silva, J. Chromatogr. A, 848 (1999) 105.
[69] P.J.M.Kwakman, D.A.Kamminga, U.A.Th.Brinkman, J. Chromatogr. 553 (1991)
345.](https://image.slidesharecdn.com/meseguer-170216182421/85/Meseguer-186-320.jpg)
![Capítulo 2. RESULTADOS OBTENIDOS Y DISCUSIÓN
163
[70] E.Orejuela, M.Silva, J. Chromatogr. A, 1007 (2003) 197.
[71] C.Molins-Legua, P.Campins-Falco, A.Sevillano-Cabeza. Anal. Chim. Acta, 378
(1999) 83.
[72] D.L. Massart, B.G.M.Vandeginste, L.M.C.Buydens, S.De Jong, P.J.Lewi,
J.Smeyers-Verbeke, Handbook of Chemometrics and Qualimetrics: Part A, Elsevier,
Amsterdam 1997.
[73] M. Valcárcel, S.Cárdenas, M.Gallego. Trends Anal. Chem., 18 (1999), 685.
[74] M. Valcárcel, S.Cárdenas, M.Gallego, Trends Anal. Chem., 21 (2002), 251.
[75] S.Igarashi, T.Nagoshi, T.Kotake, Anal. Letters, 33 (2000), 3271.
[76] L.A. Tortajada-Genaro, P.Campíns-Falcó, J.Verdú-Andrés, F.Bosch-Reig, Anal.
Chim. Acta, 450 (2001) 155.
[77] S.Meseguer-Lloret, P.Campíns-Falcó, L.A.Tortajada-Genaro, F.Blasco-Gómez,
Intern. J. of Environ. Anal. Chem., 83 (2002) 405.
[78] Y. Moliner-Martínez, S.Meseguer-Lloret, L.A.Tortajada-Genaro, P.Campíns-Falcó,
Talanta, 60 (2002) 257.
[79] N.W. Barnett, R. Bos, R.N. Evams, R.A. Russell. Anal. Chim. Acta, 403 (2000) 145.
[80] S. Igarashi, T. Nagoshi, T. Kotake, Anal. Letters, 33 (2000) 3271.
[81] M.A. Marina-Sánchez, M.E. Díaz-García, A. Sanz-Mendel. Mikrochim-Acta, 106
(1992) 227.
[82] R. Escobar, Q.X. Lin, A. Guiraum, F.F. De la Rosa. Analyst, 118 (1993) 643.
[83] HG. Beere, P. Jones, Anal. Chim. Acta, 293 (1994) 237.
[84] R. Escobar, Q. Lin, A. Guiraum, F.F. De la Rosa. Inter. J. Environ. Anal. Chem., 61
(1994) 169.
[85] B.Gammelgaard, Y.P. Liao, O. Jons, Anal.Chim.Acta, 354 (1997) 107.
[86] A. Economou, A.K. Clark, P.R.Fielden, Anal-Commun., 35 (1998) 389.
[87] M. Derbyshire, A. Lamberty, P.Gardiner, Anal. Chem., 71 (1999) 4203.
[88] Y. Xu, F.G. Bessoth, JCT Eijkel and A. Manz, Analyst, 125 (2000) 677.
[89] A.E. Greenberg, L.C. Clesceri, A.D. Eaton, Standard Methods for Examination of
Water and Wastewaters, American Public Health Association, Washington D.C., (1992).
[90] M. Stoeppler. Sampling and Sample preparation, Elsevier, Berlin, 1997.
[91] E. Nakayama, K. Isshiki, Y. Sohrin and H. Karatami, Anal. Chem., 61 (1989) 1392.
[92] A.A.Alwarthan, K.A.J. Habib, A. Townshend, Fres. J. Anal. Chem., 337 (1990) 848.
[93] B. Yan, P.J. Worsfold, Anal. Chim. Acta, 236 (1990) 287.
[94] V.A. Elron, K.S. Johnson, K.H. Coale, Anal. Chem., 63 (1991) 893.
[95] H. Obata, H. Karatani, E. Nakayama, Anal. Chem., 65 (1993) 1524.
[96] Z. Zhang, W. Qin, S. Liu, Anal. Chim. Acta, 318 (1995) 71.
[97] S. Hirata, Y. Hashimoto, M. Aihara, G. Vitharana-Mallika, Fres. J. Anal. Chem., 355
(1996) 676.
[98] K. Okamura, T. Gamo, H. Obata, E. Nakayama, Y. Nozaki, Anal. Chim. Acta, 377
(1998) 125.
[99] W. Qin, Z.J. Zhang, F.C. Wang, Fres. J. Anal. Chem., 360 (1998) 130.
[100] A.R. Bowie, E.P. Achterberg, R.F.C. Mantoura, P.J. Worsfold, Anal. Chim. Acta,
361 (1998) 189.
[101] S. Hirata, H. Yoshihara, M. Aihara, Talanta, 49 (1999) 1059.
[102] V. Cannizzaro, A.R. Bowie, A. Sax, E.P. Achterberg, P.J. Worsfold, Analyst, 125
(2000) 51.](https://image.slidesharecdn.com/meseguer-170216182421/85/Meseguer-187-320.jpg)
![REFERENCIAS BIBLIOGRÁFICAS
164
[103] C.A. Chang, H.H. Patterson, Anal. Chem., 52 (1980) 653.
[104] Q.X. Lin, A. Guirarum, R. Escobar, F.F. de la Rosa, Anal. Chim. Acta., 283 (1993)
379.
[105] A.R. Bowie, P.R. Fielden, R.D. Lowe, R.D. Snook, Analyst, 120 (1995) 2119.
[106] Y. Zhou, G. Zhu, Talanta, 44 (1997) 2041.
[107] H. Zamrow, K.H. Coale, K.S. Johnson, C.M. Sakamoto, Anal. Chim. Acta, 377
(1998) 133.
[108] P. Campíns-Falcó, L.A. Tortajada-Genaro, F. Bosch-Reig, Talanta, 55 (2001) 403.
[109] F.W.Fifield, P.J.Haines, Environ. Anal. Chem., Blackie, London, 1995.
[110] N. Shakulashvili, T. Faller and H. Engelhardt, J. Chromatogr. A, 895 (2000) 205.
[111] W. Zmijewska, H. Polowska-Motrenko, H. Stokowska, J. Radioanal. Nucl. Chem.,
116 (1987) 243.
[112] N.S. Chong, M.L. Norton, J.L. Anderson, Anal. Chem., 62 (1990) 1043.
[113] M.A. Barreiros, M.L. Carvalho, M.M. Costa, M.I. Marques, M.T. Ramos, X Ray
Spectrom., 26 (1997) 165.
[114] R. Saran, T.S. Basu-Baul, P. Srinivas, D.T. Khalthing, Anal. Lett., 25 (1992) 1545.
[115] T. Balaji, P. Chiranjeevi, G.R.K. Naidu, Anal. Lett., 31 (1998) 1081.
[116] B. Budic, Fres. J. Anal. Chem., 368 (2000) 371.
[117] K.H. Lee, M. Oshima, T. Takayanagi, S. Motomizu, Anal. Sci., 16 (2000) 731
[118] J.R. Gord, G. Gordon, G.E. Pacey, Anal. Chem., 60 (1988) 2.
[119] D.F. Marino, J.D. Ingle, Anal. Chem., 53 (1981) 292.
[120] H. Sakai, T. Fujiwara, M. Yamamoto and T. Kumamaru, Anal. Chim. Acta, 221
(1989) 249.
[121] R. Ferrer, J.L. Beltrán, J. Guiteras, Talanta, 45 (1998) 1073.
[122] J. Amador-Hernández, A. Cladera, J.M. Estela, P.L. López-Alba, V. Cerda,
Analyst, 123 (1998) 2235.
[123] F. Blasco-Gómez, F. Bosch-Reig, P. Campíns-Falcó, Talanta, 49 (1999) 155.
[124] M. Azubel, F.M. Fernández, M.B. Tudino, O.E. Troccoli, Anal. Chim. Acta, 398
(1999) 93.
[125] P. Campins-Falcó, L.A. Tortajada-Genaro, S. Meseguer-Lloret, F. Bosch-Reig,
Anal. Bioanal. Chem., 374 (2002) 1223.
[126] D. González-Robledo, M.Silva, D.Pérez-Bendito, Anal. Chim. Acta, 217 (1989)
239.
[127] P. Campins-Falcó, L.A.Tortajada-Genaro, F.Bosch-Reig, Talanta, 55 (2001) 403.
[128] R.D. Jalkian, M.B. Denton, Appl. Spectrosc., 42 (1998) 1194.
[129] A. Segura-Carretero, J. Rodríguez-Fernández, A.R. Bowie, P.J. Worsfold, Analyst,
125 (2000) 387.
[130] T.P. Burt, A.L. Heathwaite, S.T. Trudgill. Nitrate: Processes, Patterns and
Management, John Wiley & Sons, New York, 1993.
[131] E.-S. A. Badr, E.P. Achterberg, A.L. Tappin, S.J. Hill, C.B. Braungardt, Trends
Anal. Chem., 22 (2003) 819.
[132] K.Cameron, C.Madramootoo, A.Crolla, C.Kinsley, Wat. Res., 37 (2003) 2803.
V. P. Aneja, B. Bunton, J. T. Walker, B. P Malik, Atm. Environ., 35 (2001) 1949.
[133] S.E.Tsiouris, A.P.Mamolos, K.L.Kalburtji, D.Alifrangis, Agric. Ecosyst. & Envir.,
89 (2002) 117.
[134] S.H. Ensign, M.A. Mallin, Wat. Res., 35 (2001) 3381.](https://image.slidesharecdn.com/meseguer-170216182421/85/Meseguer-188-320.jpg)
![Capítulo 2. RESULTADOS OBTENIDOS Y DISCUSIÓN
165
[135] V. P. Aneja, B. Bunton, J. T. Walker, B. P Malik, Atm. Environ., 35 (2001) 1949.
[136] B.M. Jones, C.G. Daughton, Anal. Chem., 57 (1985) 2320.
[137] EPA-821-R-01-004. Method, 1687. 2001.
[138] G. Alavoine, B. Nicolardot, Analusis, 28 (2000) 88.
[139] J.H. Sharp, K.R. Rinker, K.B. Savidge, J.Abell, J.Y. Benaim, D. Bronk, D.J.
Burdige, G. Cauwet, W. Chen, M.D. Doval et al., Mar. Chem., 78 (2002) 171.
[140] A. Aminot, D.S. Kirkwood and R. Kérouel, Mar.Chem., 56 (1997) 59.
[141] M. Kaminski, D.Jastrzebski, A.Przyjazny, R.Kartanowicz, J.Chromatogr. A, 947
(2002) 217.
[142] F. Dai, V.P. Burkert, H. N. Singh, W.L. Hinze, Microchemical Journal, 57 (1997) 166.
[143] P. Campíns-Falcó, C. Molins-Legua, A. Sevillano-Cabeza and L. A. Tortajada
Genaro, J. Chromatogr. B, 759 (2001) 285.
[144] H.Mana, U.Spohn, Fres. J. Anal. Chem., 366 (2000) 825.
[145] A.Sevillano Cabeza, Y.Moliner Martinez, C.Molins Legua, P.Campíns Falcó,
Anal.Bioanal.Chem., 376 (2003) 918.
[146] J. Verdú, D.L. Massart, Applic. Spectrosc., 52 (1998) 1425.
[147] V.Centner, D.L.Massart, O.E. de Noord, S.de Jong, B.M.Vandeginste and C.Stena.
Anal.Chem., 68 (1996) 3851.
[148] J. Li, P. Purnendu, K. Dasgupta, Anal. Chim. Acta, 398 (1999) 33.
[149] D. Tang, L.Wen, P.H. Santschi, Anal. Chim. Acta, 408 (1-2) (2000) 299.](https://image.slidesharecdn.com/meseguer-170216182421/85/Meseguer-189-320.jpg)





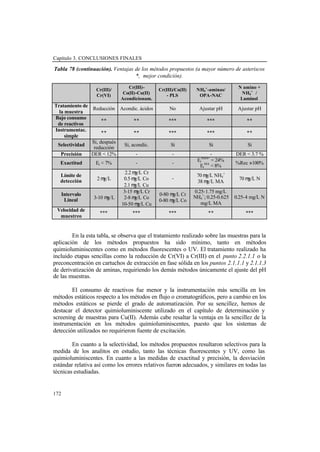

















![190
INTRODUCTION
Ammonium is one of the most widely used industrial chemicals. The principal
uses of ammonia are direct-application fertiliser, conversion to other chemical fertilizers,
fibres, explosives, plastics, and animal feeds. It is also used as food additive, in
insecticides, in the manufacture of household detergents and cleaners, and as a major
commercial refrigerant [1]. It is the most serious pollutant causing eutrophication of
aqueous environments. Although ammonium is an important link in the nitrogen cycle of
aquatic systems, its determination is still a delicate task. Its concentration in water
systems is regulated by legislation to guarantee the quality of the water supplied for
human consumption, being the limit (parametric value) established in 0.5 mg/L , and 2
mg/L when we are talking about Total Kjeldahl Nitrogen [2]. The performance
characteristic of the method of analysis is also specified in the legislation, it has to be
capable of measuring concentrations equal to the parametric value with trueness,
precision and limit of detection of 10 % of the parametric value [2].
The reference methods for ammonium determination include wet technique such
as trimetry, colorimetry (Nesslerization, Phenate or automated phenate) or ammonia-
selective electrode [3]. These methods suffer from several chemical interferences, can
require distillation step and often offer lack of sensitivity [4-7].
In order to improve selectivity some procedures have been described in the
literature in which a ion chromatography was applied. In these papers a weakly basic
anion-exchange resin column [8] or high capacity cation exchange column [9], have been
used to separate ammonium to the other inorganic cations or/and aliphatic amines which
can be present in water samples.
A number of methods exist for measuring ammonium with
Orthophthaldialdehyde (OPA)-derivatization reactions, such as OPA-sulphite reagent [5]
or more recently OPA- tioglycolate [10] and OPA-NAC [4] by forming fluorescent
derivates. Background fluorescence from naturally occurring organic compounds may not
be negligible in some fresh waters and then the results obtained by the methods could be
biased [5].
Chemiluminescence procedures have also been developed for ammonium
determination [11-12], due to its high sensitivity, rapidity, simplicity and feasibility. The
reagent mainly used is luminol combined with hypochlorite and the procedures are
systems of flow injection analysis (FIA), not being described its application by HPLC in
order to improve selectivity.
Despite the known reaction between nitrogen compounds and dansyl chloride
reagent (Dns-Cl), the reaction has not been applied to the quantitative determination of
ammonium ions with fluorimetric nor postcolumn chemiluminescence generation ( with
reagents such Bis-(2,4,6-trichlorophenyl)oxalate, TCPO) detection systems. However,
dansylation has been widely used for the determination of analytes such as biogenic
amines in plants [13] as low-molecular-mass amines applied to water samples [14-16],](https://image.slidesharecdn.com/meseguer-170216182421/85/Meseguer-214-320.jpg)
![191
phenols in surface waters [17], phenoxyl-type N-methylcarbamate pesticide residues in
fruit juices [18] and amphetamines in urine [19-20].
Based on our experience in the derivatization of amine compound with Dns-Cl
and TCPO post-column chemiluminescence generation [16, 19-20], in this work, a fast,
sensitive and selective method, has been developed for the screening and quantification of
ammonium in water samples at low concentration level. The solution derivatization of
ammonium in the sample with dansyl chloride reagent was studied, followed by HPLC in
order to separate ammonium signal from other nitrogen-compounds response.
Optimization of the separation on a C18 analytical column and postcolumn mixing with
TCPO/H2O2 was carried out. A screening procedure for aliphatic amines and ammonium
in water samples of different nature has been developed. Ammonium determination was
carried out in different kind of samples. Moreover, the method was applied to several
samples after applying Kjeldahl treatment in order to obtain values of Total Kjeldahl
Nitrogen.
EXPERIMENTAL
Apparatus
The chromatographic system used consisted of a quaternary pump (Hewlett
Packard Series 1050) equipped with a high-pressure six-port valve (Rheodyne model
7000). The volume of sample loop was 20 µl.
The post column instrument (PCX 5100), from Pickering Laboratories (Mountain
View, CA, USA), consisted of two isocratic pumps. The chemiluminescence detector was
a Jasco CL-1525 (Ishika-cho, Hachioji-shi, Tokio, Japan) which was coupled in series
and linked to a data system Borwin-chromatography software and the interface Hercule
lite (JMBS, France) used for data acquisition and storage. The model CL-1525 detector is
equipped with JASCO´s uniquely designed coiled PTFE tube flow cell placed
inmediatelly next to a highly sensitive end-on photomultiplier. All the assays were carried
out at room temperature.
For sample Kjeldahl treatment, the Digestion Unit was a Tecator Digestion
system 6, 1007 Digestor (Höganäs, Sweden), and the Distillation Unit was a Büchi 323
Distillation Unit (Switzerland).
Reagents
All the reagents were of analytical grade. Stock standard solutions of ammonium
were prepared by dissolving ammonium chloride (Probus, Barcelona, Spain) in nanopure
water (1000 mg/L). Acetonitrile (J.T.Baker, Deventer, Holland), Tetrahydrofuran (Fluka,
Steinheim, Switzerland), Methanol and Acetone (Scharlau, Barcelona, Spain) were of
HPLC grade. Methylamine (MA), Ethylamine (EA), N-Propylamine (N-Pra), Butylamine
(BA), Dimehylamine (DMA), Diethylamine (DEA), Pentylamine (PeA), Hexylamine
(HA) and Dansyl Chloride (Dns-Cl) were obtained from Sigma (St.Louis, MO). TCPO,
bis (2, 4, 6-trichlorophenyl oxalate), tetrahydrofuran and Imidazole (99%) from Fluka](https://image.slidesharecdn.com/meseguer-170216182421/85/Meseguer-215-320.jpg)
![192
(Steinheim, Switzerland), and H2O2 (30%), sodium carbonate and sodium hydroxide from
Merck (Darmstadt, Germany) were also used.
Sodium thiosulphite and potassium sulphate from Prolabo (Fontenay, France),
sodium hydroxide, sulphuric acid and sodium carbonate from Merck (Darmstadt,
Germany), and red mercury oxide from Panreac (Barcelona, Spain) were used for sample
Kjeldahl treatment.
Columns and Mobile -Phases
A LichroCART RP 18 (125x4 mm i.d., 5 µm particle φ) (Merck, Darmstadt,
Germany) column was used for separation of ammonium derivative from reagent excess
and from other nitrogen-compound derivatives. An acetonitrile:imidazole solution (1
mM, pH=7.0) (50:50 v/v) mixture in gradient elution mode was used as eluent at flow
rate of 1 ml/min.
The mobile phase gradient used was the following: gradient elution mode 1:
acetonitrile:imidazole 50:50 at zero time, 70:30 at 6 min, and 50:50 at 7 min. When
amine standards were studied, the gradient elution mode was extended in order to assure
the elution of all amine derivatives following the gradient elution 2, established in our
previous studies [15]: acetonitrile:imidazole 50:50 at zero time, 90:10 at 9 min, and 50:50
at 10 min.
All the solvents were filtered with a 0.45 µm nylon membrane (Teknokroma,
Barcelona, Spain) and degassed with Helium before use.
Preparation of solutions
Standard solutions of ammonium were prepared by dissolving the pure
compounds in water (1000 µg/ml). Working ammonium solutions were prepared by
diluting the standard solutions in water. Dns-Cl 5 mM solution was prepared by
dissolving the pure compound in acetonitrile. All solutions were stored in the dark at 4
ºC.
Solution derivatization of ammonium
Ammonium was derivatized according to the method described by Marce et al.
[13] with some modifications. To 1.55 ml of ammonium standard, 0.05 ml of 200 mM
carbonate buffer (pH 9), 0.28 ml of acetonitrile and 0.12 ml of 5 mM dansyl chloride (in
acetonitrile) were added successively, and the mixture was incubated at 70 ºC for 10 min.
An aliquot (20 µl) of the solution was injected into the HPLC system.](https://image.slidesharecdn.com/meseguer-170216182421/85/Meseguer-216-320.jpg)

![194
bottled water (S1), tap water (S2), fountain water (S3), irrigation ditch water (S4)(S5)
lake water (S6), sea water (S7) (S8) (S9), residual water from a factory (S10). Water
samples were collected, filtered through 0.45 µm and acidified to pH 2 with HCl.
Previously to the analysis, the samples were alkalised with NaOH to pH 6-7.
Depending on the ammonium concentration in the sample, sample volumes
ranged between 0.15 to 1.5 ml were taken. For bottled, tap, irrigation ditch and sea water
1.5 ml samples were processed. Lower volumes were taken for fountain (0.643 ml) and
lake water samples (0.188 ml). For residual water sample 0.12 ml of 1:10 sample dilution
was employed. The samples were also spiked with known amounts of the stock standard
solutions of ammonium to give added concentrations of 0.15 and 0.3 mg/L. In all cases
the volume of sample or sample plus spike was completed if necessary to 1.55 mL with
deionized water.
These volumes were processed according to the procedure described above for
standard solutions.
Kjeldahl treatment
Kjeldahl treatment was applied to 100 ml of fountain water (S3), irrigation ditch
water (S4), lake water (S6) and sea water (S9). For the application of Kjeldahl treatment
[21], 6.7 g of potassium sulphate, 0.52 g of red mercury oxide and 10 ml of concentrated
sulphuric acid, were added to 100 ml of sample in a digestion tube. Then, the tubes were
placed on Kjeldahl digestor, which was previously heated at 370ºC. Heating was running
until white fumes appeared and then, digestion continued for 30 minutes more. After that,
digestion residue was cooled and diluted up to 100 mL with nanopure water. Distillation
was performed by adding 35 ml of distillation reagent (25 g of sodium thiosulphate and
500 g of sodium hydroxide diluted up to 1L with nanopure water). Distilled ammonia was
picked up on 25 ml of 0.04 N sulphuric acid until 80 mL. This solution was adjusted to
pH 6-7 and diluted up to 100 mL. 1.55 mL of the resulted solution were processed
according to the procedure described above for standard solutions.
RESULTS AND DISCUSSION
Optimization of precolumn ammonium dansylation
Ammonium was derivatized according to the conditions established by Marcé et
al.[13] for polyamines, with some modifications. For establishing the optimal conditions
the dansylated derivatives were injected in the HPLC systems and the post column
chemiluminescence generation with TCPO was performed according to the procedure
described by our research group for aliphatic amines [16] (see experimental sectionand
Figure 1) in order to register the chromatograms. The dansylated ammonium eluted at
2.4 min (Figure 2).](https://image.slidesharecdn.com/meseguer-170216182421/85/Meseguer-218-320.jpg)

![196
FIGURE 3. Influence of solvent volume, employed in the reaction derivatization with dansyl
chloride (Conditions: DnsCl 0.45 mM), and reagent concentration (Dns-Cl) (Conditions:
Acetonitrile Volume 0.4 mL) on the blank signal.
The reagent concentration (Dns-Cl) in the mixture reaction was also studied in
the range 0.15-0.75 mM maintaining constant the acetonitrile volume at 0.4 mL. 0.3 mM
was chosen as optimum reagent concentration because provided a low blank signal
(Figure 3) without losing in ammonium response.
According to these results the optimal conditions were those shown in Table 1. In
this table are compared the conditions established by Marcé et al.[13] and those
optimized in this paper, as can be seen the original procedure has been simplified in order
to make it quicker.
TABLE 1. Comparison of the derivatization procedure for solution derivatization according
to Marcé et al and for that proposed in this paper.
Marcé et al. Procedure[13] Procedure optimized in this work
200 ì L sample/standard + 40 ìL internal
standard + 200 ìL saturated sodium carbonate
+ 400 ì L DnsCl 5 mg/ml in acetone
1.55 mL sample/standard + 50 ìL carbonate
buffer 0.2 M pH 9 + 400 ìL DnsCl 1.5 mM in
acetonitrile
Incubate in the dark overnight at room
temperature or for 10 min at 70ºC
Incubate 10 minutes at 70ºC
Add 100 ìL of proline solution (100 mg/ml)
and incubate in the dark for 30 minutes
Inject 20 ì L onto the HPLC system
Extract dansylated polyamines in 500 ìL of
toluene and mix for 30 seconds
Remove 400 ìL of organic phase, dry, and
redissolve with 800 ìL of acetonitrile
Pass the sample through a 0.45 ìm pore size
syringe filter
Inject 20 ì L onto the HPLC system
0
10000
20000
30000
40000
50000
60000
70000
80000
0 0.2 0.4 0.6 0.8 1
Acetonitrile Volume (mL) or DnsCl
Concentration (mM)
BlankPeakHeigh(V)
Variable Acetonitrile Volume
Variable DnsCl concentration
Optimum conditions](https://image.slidesharecdn.com/meseguer-170216182421/85/Meseguer-220-320.jpg)
![197
Analytical performance characteristics
Table 2 shows the calibration graph obtained in the best conditions. Good
linearity was obtained between 0.027-0.75 mg/L of ammonium and the detection limit
established as the concentration required to generate a signal-to-noise ratio of 3, was 8
µg/L. The chromatogram corresponding to detection limit appears in Figure 4. The
achieved detection limit is far away from legislation limit established (0.5 mg/L for non
digested samples, and 2 mg/l for samples treated by Kjeldahl method).
TABLE 2. Equations of the calibration graphs obtained in several conditions. The analytical
signal used was peak height, except (*) where area instead of height was employed.
Condition
Dns-Cl
(mM)
NaCl
Calibration curve
Y = (a ±± sa) + (b ±± sb)· C
(n, r2
,sy/x)
Detection
Limit
(µg/L)
Linear
Interval
(mg/L)
[1] 0.15
Y = (12 ± 6) + (130 ± 10)· C
(5, 0.9814,9)
60 0.28-1
[2] 0.3
Y = (2 ± 7) + (960 ± 30)· C
(10, 0.9935, 16)
Y* = (3 ± 90) + (12300 ± 400)· C
(10, 0.9929, 205)
8 0.027-0.75
[3] 0.3 3.5 g/l
Y = (14± 5) + (930 ± 30)· C
(3, 0.9998, 3)
8 0.027-0.75
[4] 0.3 35 g/l
Y =(19 ± 14) + (980 ± 70)· C
(3, 0.9942, 16)
8 0.027-0.75
0 2 4 6 8 10
ChemiluminescenceSignal
Time (min)
NH4+
R
MA
EA
N-PRA + DMA
BA
DEA PEA
HA
R
1
2
3
LD NH4+
FIGURE 4. Chromatograms
corresponding to 1: blank, 2:
ammonium standard of 8 µg/L
and 3: mixture of amines:
Methylamine (MA) 0.1 mg/L,
Ethylamine (EA) 0.2 mg/L, N-
Propylamine (N-PrA) 0.3 mg/L,
Dimethylamine (DMA) 0.3 mg/L,
Butylamine (BA) 0.3 mg/L,
Diethylamine (DEA) 0.5 mg/L,
Hexylamine (HA) 0.3 mg/L and
ammonium (NH4+) 1.5 mg/L.](https://image.slidesharecdn.com/meseguer-170216182421/85/Meseguer-221-320.jpg)
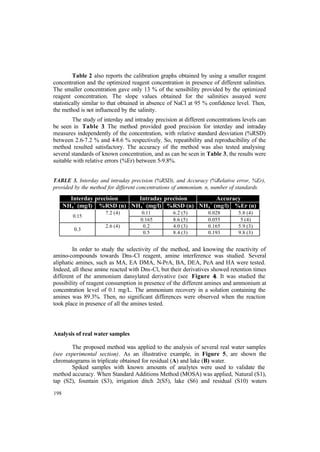
![199
presented similar slopes to that obtained by the calibration graph with standards (see
Table 2) at 95 % confidence level as t tests [22] given in Table 4 show, therefore no
matrix effect was found. For sample S5 (irrigation ditch 1) both slopes, MOSA and
ammonium standard equation, were similar at 98 % confidence level. For the sea waters
only S7 provided similar slopes at 99 % confidence level, while S8 and S9 gave matrix
effect as can be seen in Table 4. When matrix effect is present, ammonium concentration
must be calculated by applying MOSA. The MOSA equations (ordinate: a(sa), slope:b(sb),
r2
, n, sy x) for S8 and S9 samples were : 57(5), 490(30), 0.9976, 4, 8.8 and 55(5), 320(30),
0.9707, 5, 8.6, respectively. The MOSA slopes were smaller than that corresponding to
the ammonium standard calibration graph (Table 2).
TABLE 4. t test for the comparison of the slopes of the ammonium standard calibration
graph and of the MOSA equations for all samples tested. The tabulated values correspond to
95 % confidence level except * (98 %) and **(99 %).
Sample Calculated
F
Tabulated
F
Conclusión 1 Calculated
t
Tabulated
t
Conclusión
2
S1-Bottled 3.988 17.443 Homogenous
variances
1.086 2.7767 Similar
slopes
S2-Tap 6.150 799.5 Homogenous
variances
2.940 3.1824 Similar
slopes
S3-Fountain 3.205 864.16 Homogenous
variances
0.614 2.7764 Similar
slopes
S4-Irrigation
ditch 1
1.606 864.16 Homogenous
variances
3.540 2.7764
3.7469*
Similar
slopes
S5-Irrigation
ditch 2
92.003 17.443 Non
homogenous
variances
2.024 12.603 Similar
slopes
S6-Lake 44.31 17.443 Non
homogenous
variances
1.264 12.496 Similar
slopes
S7-Marine 1 2.23 799.5 Homogenous
variances
3.894 3.1824
5.84**
Similar
slopes
S8-Marine 2 5.36 17.443 Homogenous
variances
5.960 2.7764
4.6041**
Matriz
effect
S9-Marine 3 5.421 17.443 Homogenous
variances
8.137 2.7764
4.6041**
Matriz
effect
S3- Kjeldahl 1.659 647.79 Homogenous
variances
0.071 4.3027 Similar
slopes
S4- Kjeldahl 2.378 16.044 Homogenous
variances
0.057 2.5707 Similar
slopes
S6- Kjeldahl 2.726 647.79 Homogenous
variances
2.311 4.3027 Similar
slopes
S9- Kjeldahl 3.409 864.16 Homogenous
variances
0.619 2.7764 Similar
slopes
S10-
Kjeldahl
1.571 799.5 Homogenous
variances
1.179 3.1824 Similar
slopes](https://image.slidesharecdn.com/meseguer-170216182421/85/Meseguer-223-320.jpg)

![201
FIGURE 6. Typical chromatograms for the fountain water (0.643 mL of sample to 1.55 mL)
fortified with 0, 0.15 and 0.3 mg/L of ammonium. n in the last column is the number of
solutions used for applying MOSA.
In Table 6 is described the screening response of the proposed method to all
samples assayed taking as reference the concentration limit established by legislation. As
can be seen, ammonium was found in S3, S6, S7, S8, S9 and S10, but only residual water
(S10) and Marine water (S7) exceed the limit established by legislation (0.5 mg/L). Table
6 also shows the ammonium content found by using the calibration graph of ammonium
(see Table 2) and MOSA method providing similar results. Fountain and residual waters
were also analysed by using OPA-NAC method [4]. The results were: 0.37(s=0.02, n=3)
and 32(s=0.4, n=3), respectively. Both values can be considered statistically equal (at a
confidence level of 95 % ) to those obtained by the proposed method. The quantified
ammonium in residual water should be observed with reserve due to the high dilution
carried out in the sample for both chemiluminescence and fluorescence methods.
A representative group of these samples were treated to evaluate Total Kjeldahl
Nitrogen (TKN). As t tests given in Table 4 indicate the slope of the MOSA method is
similar to that corresponding to the standard calibration graph of ammonium. Therefore
neither of the samples presented matrix effect even sea water and the ammonium
concentration in the samples was obtained directly from the calibration graph with
standards of ammonium (Table 5). The samples were also spiked with known amounts of
ammonium in order to validate the accuracy and the found ammonium concentrations are
shown in Table 4. As can be seen, low relative errors between 0.3 and 10.7% were
achieved.
0 2 4 6
ChemiluminescenceSignal
Time (min)
S3
S3 + 0.15 mg/l NH4+
S3 + 0.3 mg/l NH4+
NH4+
NH4+
NH4+](https://image.slidesharecdn.com/meseguer-170216182421/85/Meseguer-225-320.jpg)

![203
three assayed marine water samples required the application of standard additions
method for the quantification of ammonium due to the presence of matrix effect but not
when the Kjeldahl treatment is applied.
Acknowledgements
The authors are grateful to the Ministerio de Ciencia y Tecnología of Spain for
financial support received for the realization of Project BQU2003-06138 . Susana
Meseguer Lloret expresses her gratitude to Ministerio de Educación y Cultura (Spain) for
the predoctoral grant.
Bibliographic References
[1] http:/www.atsdr.cdc.gov/es/toxfaqs/es_tfacts33.pdf
[2] European Directive 98/83/CE.
[3] Díaz-Santos S.A. Métodos Normalizados para el Análisis de Aguas Potables y
Residuales. Editorial Apha-Awwe-WPCF. 17 Ed. (1992).
[4] S.Meseguer Lloret, C.Molins Legua, P.Campins Falcó, Intern.J.Environ.Anal.Chem.,
82 (2002) 475-489.
[5] A. Aminot, R.Kerouel, D.Birot, Wat.Res. 35-7 (2001) 1777-1785
[6] J.P. Hart, A.K. Abass, D.C. Cowell, A. Chappel, Electroanalysis, 11(1999) 406-411
[7] J. Schwarz, H. Kaden, G. Pausch, Fresenius J. Anal. Chem.., 367(2000) 396-398.
[8] M-Mori, K.Tanaka, M.I.H.Helaleh, Q.Xu, M.Ikedo, Y.Ogura, S.Sato, W.Hu,
K.Hasebe, J.Chromatography A, 997 (2003) 191-197.
[9] D.H.Thomas, M.Rey, P.E.Jackson, J.Chromatography A, 956 (2003) 181-186.
[10] H.Mana and V.Spohn, Fresenius J Anal Chem, 366, 825-829 (2000).
[11] J.Li, P.K.Dasgupta, Anal.Chim.Acta., 398 (1) (1999) 33-39
[12] W.Qin, Z.J.Zhang, B.X.Li, Y.Y.Peng, Talanta 48 (1999) 225-229.
[13] M.Marcé, D.S.Brown, T.Capell, X.Figueras, A.F.Tiburcio, J. Chromatog. 666
(1995) 329-335.
[14] M.Cobo, M.Silva, J. Chromatog. A, 848 (1999) 105-115.
[15] S. Meseguer Lloret, C. Molins Legua and P. Campins Falco, J. Chromatog. A, 978
(2002) 59-69.
[16] S. Meseguer Lloret, C. Molins Legua, J. Verdú Andrés, P. Campins Falco, J.
Chromatog. A, in revision.
[17] P.J.M.Kwakman, D.A.Kamminga, U.A.Th.Brinkman, J. Chromatog. 553 (1991)
345-356.
[18] E.Orejuela, M.Silva, J. Chromatog. A, 1007 (2003) 197-201.
[19] C.Molins-Legua, P.Campins-Falco, A.Sevillano-Cabeza. Anal. Chim, Acta. 378
(1999) 83-93.
[20] C.Molins-Legua, P.Campins-Falco, A.Sevillano-Cabeza, Analyst. 123 (1998) 2871-
2876.
[21] Application Note ASN 52/85. Tecator Manual Digestion system 1007 (1985).
[22] D.L. Massart, B.G.M. Vandeginste, L.M.C. Buydens, S. De Jong, P.J. Lewi y J.
Smeyers-Verbeke. Handbook of Chemometrics and Qualimetrics: Part A, Elsevier,
Amsterdam 1997.](https://image.slidesharecdn.com/meseguer-170216182421/85/Meseguer-227-320.jpg)


![206
1. Introduction
The chemiluminescence technique provides methods for trace analysis that are
attractive because of their high sensitivity and fast kinetics. This technique, combined
with FI systems provides good accuracy and precision.
Peryoxalate chemiluminescence reaction (TCPO/H2O2 system), has been
extensively used in recent years for liquid chromatography of fatty acids [1], synthesis,
characterisation and analytical evaluation of oxamide reagents [2], determination of
amphetamines in urine samples [3], inmunoassays [4], detection of proteins separated by
capillary zone electrophoresis [5], resolution of carboxylic acids by HPLC [6],
determination of cholesterol [7], detection of retinoids using normal-phase
chromatography [8] and the study of emission of spectroscopic properties of water
soluble porphyrins with d- and f- electron metals [9, 10]. The use of porphyrins can be
remarked by their high selectivity to metal ions [10].
Metal ions are usually introduced into water cycle by industrial dumping and
many of them present toxic effects for human life; therefore, their maximum tolerated
concentration have been fixed by legislation, being for copper(II) 50 µg/l [11]. In order to
control these values in real water samples, fast automatic systems are required; these type
of systems are called screening systems [12, 13], and provide a binary yes/no response
that let to distinguish samples containing Cu(II) over or under legislation level with good
reliability, fast speed, versatility and low cost.
Igarashi et al. [10] proposed an static method for Copper (II) determination based
on coproporphyrin I-Cu(II)/TCPO/H2O2 chemiluminescence reaction. Based on their
studies, we are proposing in this paper an automatic FI method for the screening of Cu(II)
in water samples.
2. Experimental
2.1 Apparatus
Flow injection chemiluminescence measures were carried out with a FireFly
Chemiluminescence Detector (Global FIA Inc. P.O. Box 480, Fox Island, WA 98333)
designed specially for use in flow through systems. Light generated in a liquid core
waveguide is internally reflected and transmitted to a small but powerful photo multiplier.
FI registers were monitored by an Hercule Lite-Chromatography interface connected on
line to a personal computer.
The flow system consisted of two Gilson Minipuls 3 peristaltic pump furnished
with poly(vinyl chloride) pumping tubes, two Rheodyne (Cotati, CA) 5041 injection
valves, PTFE tubing of 0.5 mm i.d., and a water bath provided by a heater magnetic
stirrer (Raypa, Spain).](https://image.slidesharecdn.com/meseguer-170216182421/85/Meseguer-230-320.jpg)

![208
CTAC/H2O2 stream (at 0.7 ml/min) in the second 200 µl coil to fill the 50 µl sample loop.
Pump 2 consisted of two water carrier streams. One of them at 5 ml/min passes through
sample loop(IV1), and the other at 2.9 ml/min passes through TCPO loop(IV2), previously
filled by means of a syringe. Injection valve positions were activated manually. When
injection valves were in position 1 (loading), only nanopure water was reaching the
chemiluminescence detector. Both injection valves must be switched to position 2
(injection) at the same time, in order to allow both sample and reagents plugs reaching
detector simultaneously. The height of the FI signal obtained must provide the binary
(yes/no) response.
FIGURE 1: FI system designed for the screening of water samples for copper (II) using a
quenching chemiluminiscence determination. IV, injection valve; CLD, chemiluminiscence
detector.
3. Results and discussion
3.1. Checking the FireFly Chemiluminescence Detector
The detector was checked following the FI procedure described by L.A. Tortajada
et al. [14] for Cr(III)/luminol/H2O2 reaction. Table 1 shows log-log calibration curves and
detection limits obtained by this investigation group in recent publications [14, 15, 16]
using a Hitachi F-4500 spectrofluorimeter and the same obtained with this new detector.
IV1
IV2
CLD
PUMP 1
(ml/min)
PUMP 2
(ml/min)
Standard/Sample
(10
-6
M Porphyrin)
CTAC 10
-2
M
H2O2 0.7 M
H2O
H2O
Water bath
100ºC
400 µl 200 µl
200 µl
50 µl
50 µl
TCPO
0.7
2.2
5
2.9 L = 7.5 cm
L = 12 cm](https://image.slidesharecdn.com/meseguer-170216182421/85/Meseguer-232-320.jpg)
![209
TABLE 1: Comparison of sensitivity of detectors for Cr(III)/luminol/H2O2
chemiluminescence reaction.
Reference (loga ±± sloga) (b ±± sb)
Detection
limit (µµg/l)
Detector
[14] (1.91 ± 0.03) (1.19 ± 0.03) 1.3 Hitachi F4500
[15] (1.97 ± 0.07) (1.25 ± 0.07) 1.2 Hitachi F4500
[16] (1.84 ± 0.03) (1.16 ± 0.03) 1.6 Hitachi F4500
This work (1.04 ± 0.06) (1.25 ± 0.06) 1.4 FireFly CLD
3.2. Optimization of the flow system
Emission chemiluminescence spectrum was obtained for a blank solution
following static conditions (1.12 µM coproporphyrin I, 0.0096 M CTAC, 1.795 mM
TCPO and 0.743 M H2O2). In Figure 2a) it can be seen that there is an emission
maximum at 622 nm. Reaction kinetics curve at 622 nm was also obtained. Figure 2b)
shows that the kinetics of this reaction was very fast, it took place in about 10 seconds.
FIGURE 2: a) Emission chemiluminescence spectrum for a blank. b) Reaction Kinetic curve.
Conditions: 1.12· 10
-6
M coproporphyrin I, 0.0096M CTAC, 0.743 M H2O2, 1.795 mM TCPO.
Optimization of the flow system was carried out with a blank solution using only
pump 2 and injecting both coproporphyrin I 10-5
M/H2O2 variable/CTAC variable/borax
0.1M pH 7.4 (IV1, loop 160 µl) and variable TCPO (variable ratio MeCN:THF) (IV2,
loop 200 µl) with a syringe (see Figure 1).
Table 2 shows the variables optimized, the range studied on each case and the
optimum variable selected. Firstly, the addition of CTAC was studied based on previous
0
20
40
60
80
100
0 50 100
Time (seconds)
ChemiluminescenceSignal
b)
0
0.2
0.4
0.6
0.8
1
400 500 600 700
λλ (nm)
ChemiluminescenceSignal
a) Maximum 622 nm](https://image.slidesharecdn.com/meseguer-170216182421/85/Meseguer-233-320.jpg)
![210
studies [10]. Hydrogen peroxide and TCPO concentrations were varied according to
recent studies of the chemiluminescence reaction [2, 10]. These two reagents must reach
the detector separately in order to achieve the maximum sensitivity.
TABLE 2: Optimization of the chemical and physical variables of the FI system.
Variable Range studied Optimum
condition
CTAC conc
2· 10-5
– 9· 10-2
M 1· 10-2
M
H2O2 conc 0.1 – 1 M 0.7 M
TCPO conc 1 – 10 mM 5 mM
MeCN:THF ratio 0%:100% – 100%:0% 75%:25%
pH 4.7-10.3 7.4
Flow rate Pump 2 10 - 48 rpm 40 rpm
Loop Volume 50 – 500 µl 50 µl
Length from injection
valve (IV) to CLD
LIV1-CLD= 12 - 24 cm
LIV2-CLD=7.5 - 15 cm
LIV1-CLD= 12 cm
LIV2-CLD=7.5 cm
Length liquid core
waveguide
17 – 50 cm 17 cm
Coproporphyrin-Cu(II)
Reaction time
1 – 10 min 5 min
TCPO reagent was prepared in acetonitrile:tetrahydrofuran at different ratios
from 0:100 to 100:0. Best results were obtained for 75% acetonitrile:25% tetrahydrofuran
avoiding TCPO destruction and obtaining the best sensitivity.
CTAC/H2O2 reagent pH was studied using different buffer solutions at 0.1 M
concentration: pH 4.7 was studied in acetic/acetate buffer, pH 6.5 and 10.3 were studied
with carbonate/bicarbonate buffer and pH 7.4 was studied with borax buffer. Best results
were obtained at pH 7.4 with borax buffer.
Concerning the flow conditions, flow rate in pump 2 was varied from 10 to 48
rpm, and 40 rpm was selected as optimum, providing 5 ml/min for coproporphyrin I
stream and 2.9 ml/min for TCPO stream. Loop volume was also studied, and 50 µl were
selected because signal was more reproducible and pressure problems were avoided.
Due to the fast speed of the chemiluminescence reaction, another parameters
were optimised: distance from injection valves to chemiluminescence detector and length
of the liquid core waveguide. The length from injection valves (IV1 and IV2) to
chemiluminescence detector (CLD) (see Figure 1) was selected as the minimum possible.
This condition avoided reagent dispersion and improved sensitivity 10 times. The length
of the liquid core waveguide was also selected as the minimum possible for the detector,
and it improved the sensitivity about 2.5 times.](https://image.slidesharecdn.com/meseguer-170216182421/85/Meseguer-234-320.jpg)




![215
4. Conclusions
The proposed method can be used as an automatic system for screening of Cu(II)
in water samples providing good results in terms of sensitivity, repeatability,
reproducibility, precision, accuracy and selectivity. It allows filtering sample set of study
with good reliability, low cost, versatility, speed and minimization of operator errors,
providing the analytical information of client interest.
Acknowlegements
The authors are grateful to the Ministerio de Ciencia y Tecnología for financial
support. S.M.Ll. expresses her gratitude to Ministerio de Educación y Cultura (Spain) for
the predoctoral grant. Financial support from the Spanish DGICyT (Grant BQU2001-
1815) is gratefully acknowledged.
Bibliography
[1] Y. Ohba, N. Kuroda, K. Nakashima. Anal. Chim. Acta, 465 (2002) 101-109.
[2] N.W.Barnett, R.Bos, R.N.Evams, R.A.Russell. Anal. Chim. Acta, 403 (2000) 145-
154.
[3] C.Molins-Legua, P.Campins-Falcó, A.Sevillano-Cabeza. Anal. Chim. Acta, 378
(1999) 83-93.
[4] H.A.H.Rongen, R.M.W.Hoetelmans, A.Bult, W.P.Van Bennecom. J. Pharm. Biomed
Analysis, 12 (1994) 433-462.
[5] T.Hara, J.Yokogi, S.Okamura, S.Kato, Riichiro. J. Chromatogr. A, 652 (1993) 361-
367.
[6] T.Toyo’oka, M.Ishibashi, T.Terao. J. Chromatogr. A, 627 (1992) 75-86.
[7] J.H.Mike, T.J.Cleland. Anal. Chim. Acta, 259 (1992) 73-78.
[8] P.D.Bryant, A.C.Capomacchia. J. Pharm. Biomed. Analysis, 9 (1991) 855-860.
[9] K.Staninski, M.kaczmarek, S.Lis, M.Elbanowski. J. Solid State Chem., in Press,
Corrected Proof.
[10] S.Igarashi, T.Nagoshi, T.Kotake. Anal. Letters, 33 (2000), 3271- 3283.
[11] Real Decreto 928/88 legislación española.
[12] M. Valcárcel, S.Cárdenas, M.Gallego. Trends Anal. Chem., 18 (1999), 685-694.
[13] M. Valcárcel, S.Cárdenas, M.Gallego. Trends Anal. Chem., 21 (2002), 251-258.
[14]L.A.Tortajada-Genaro, P.Campins-Falcó, J.Verdú-Andrés, F.Bosch-Reig. Anal.
Chim. Acta 450 (2001) 155-173.
[15] S.Meseguer-Lloret, P.Campíns-Falcó, L.A.Tortajada-Genaro, F. Blasco-Gómez. Int.
J. Environ. Anal. Chem., 83 (2003) 405-416.
[16] Y.Moliner-Martínez, S. Meseguer-Lloret, L.A.Tortajada-Genaro, P.Campíns-Falcó.
Talanta 60 (2003) 257-268.](https://image.slidesharecdn.com/meseguer-170216182421/85/Meseguer-239-320.jpg)








![224
INTRODUCTION
Ammonium is a micronutrient in water systems which is an important link in the
nitrogen cycle of aquatic ecosystems. However, its determination is still a delicate task,
due to it is high susceptibility to contamination, and the classic methods does not appear
to be well controlled in many laboratories [1]. Ammonium can be found in superficial,
subterranean or marine waters at low concentrations, about 10 µg/L. In residual waters it
can be found at high concentrations, about 30 mg/L due to ammonification and nitrate
reduction processes. Often, it can be found together with short-chain aliphatic primary
amines (mainly methylamine) because they are widely distributed in the environment.
Aliphatic amines are used in several chemical and manufacturing industries and are also
common components of biological systems as degradation products of organic material
such as amino acids and proteins. Besides hygienic problems due to the stinging smell,
these compounds may be hazardous to human health. In addition, they can react with
certain nitrogen-containing compounds to form nitrosamines, which are potentially
carcinogenic substances [2]. Consequently, there is an increasing interest in the
determination of aliphatic amines in various aqueous matrices. The tolerable limits for
amines are regulated as Kjeldahl Nitrogen (which include ammonium and organic
nitrogen), being between 15 and 85 mg/L for residual waters and 1 mg/L for tap water.
Liquid Chromatography combined [3] or not with derivatization [4], is generally
used for the analysis of ammonium and aliphatic amines in aqueous media. Unbiased
UV-vis or fluorimetric methods are not found in the literature, which could be used as
screening sample methods in order to reduce costs and saving time in the environmental
laboratory or in situ determinations. The Roth´s fluorimetric method [5-7] for the
determination of primary amino compounds by use of a thiol and o-Phthaldialdehyde
(OPA) as derivatizing reagents could be adapted to the determination of ammonium and
total primary amine groups. In previous studies, we have proposed the use of OPA and
the thiol N-acetyl-cisteine (NAC) reagents for the unbiased fluorescent ammonium
determination in water samples [9] exciting at 415 nm. Some advantages over ammonium
reference methods, such as Nessler reagent method or the ammonium selective electrode
method, are the lower detection limit, the absence of any systematic error and amines
interference and less toxicity of reagents. Other methods exist for measuring ammonium
with (OPA)-derivatization reactions, such as OPA-sulphite reagent [10] or OPA-
tioglycolate [11] by forming fluorescent derivatives.
Recently we have reported the determination of primary amine groups in water
samples, by performing amine derivatization on C18 cartridges and using OPA-NAC
reagents [12] and 333 nm as excitation wavelength. The response of several primary
amino groups in the OPA-NAC reaction was demonstrated similar in [12], so it was
possible to estimate the total primary amine concentration in water employing the
calibration graph of methylamine as model compound. By using this solid phase assisted
derivatization procedure, ammonia concentrations up to 1.5 mg/L was not an interference
species.](https://image.slidesharecdn.com/meseguer-170216182421/85/Meseguer-248-320.jpg)

![226
of amines: 0.75 mg/L EA and 0.75 DEA; 0.5 DMA, 0.5 N-PrA and 0.5 IPA; 0.75 BA and
0.75 PeA; 0.75 β-FEA and 0.75 HA were also used.
Nessler method [13]: Sodium-potassium tartrate solution (Panreac, Spain) was
prepared by dissolving 50g of the compound in 100 ml of nanopure water and Sodium
hydroxide (Panreac, Spain) by dissolving 12g of the compound in 50 ml of nanopure
water. Nessler reagent (NR) was prepared by dissolving 3g of mercury chloride (Merck,
Germany) in the minimum amount of nanopure water; 10 g of potassium iodine
(Guinama, Spain) was diluted up to 50 ml, and then it was added to mercury chloride
solution until a red precipitate appears; then the solution was decanted; 18g of sodium
hydroxide was diluted up to 100 ml and 40 ml of this solution were added to the HgI4
2-
solution; the final solution was diluted up to 100ml.
Kjeldahl treatment: For sample or standard digestion, red mercury oxide
(Panreac, Barcelona, Spain), potassium sulphate (Prolabo, Fontenay, France) and
sulphuric acid (Fluka Chemie, Steinheim, Switzerland) were used. Distillation reagent
was prepared by dissolving 25 g of sodium thiosulphate (Prolabo, Fontenay, France) and
500 g of sodium hydroxide (J.T.Baker, Deventer, Holland) together in 1L of nanopure
water.
Procedures
Standard solutions
OPA/NAC reaction
In a quartz cuvette were placed variable volumes of ammonium and/or amine
(alone or mixed) stock solution and nanopure water if necessary up to a volume of 1 mL
, 0.1 mL of borate buffer 0.5 M (pH= 10.8) and 0.9 mL of OPA/NAC reagent. The
reaction was assumed to start after the addition of the last drop of OPA/NAC reagent.
Each experiment was assayed by recording spectra emission between 430-600 nm (λ exc=
415) and 380-610 nm (λ exc = 333) - over the reaction time ranged 0-300 seconds. Signal
was obtained at 120 seconds for λex = 333 nm λ em = 440 nm, and at 300 s for λex = 415
nm λ em = 485 nm. All measurements were performed at 25ºC. The ammonium and
methylamine concentrations were ranged from 0 to 1.75 mg/L and 0-0.625 mg/L
respectively, following a 52
design (Table 1).
TABLE 1. Code and concentration (mg/L) of standard calibration set (5
2
design).
NH4
+
MA
(mg/L)
0 0.25 0.375 0.5 0.625
0 00 01 02 03 04
0.25 10 11 12 13 14
0.75 20 21 22 23 24
1.25 30 31 32 33 34
1.75 40 41 42 43 44](https://image.slidesharecdn.com/meseguer-170216182421/85/Meseguer-250-320.jpg)
![227
Nessler reaction
In a plastic cuvette were placed volumes varied of ammonium stock solution, 10
µL of sodium-potassium tartrate (0.177 M) and water up to constant volume (2.4 mL), 0.1
mL of NaOH 6M and 0.1 mL of Nessler reagent were added to the mixture. The reaction
was assumed to start after the addition of the last drop of reagent. Each experiment was
assayed by recording spectra between 300-700 nm at 30 s intervals over the reaction time
ranged 0-600 seconds. Signal was obtained at 425 nm and 400 seconds. Absorbance
signal was measured against water blank. All measurements were performed at 25ºC.
Kjeldahl treatment
For the application of Kjeldahl treatment [14], several digestion-distillation
conditions were assayed (see Table 2). In the digestion step, potassium sulphate amount
was varied in the range 2.22-10.5 g, mercury oxide between 0.0332-0.525 g and
concentrated sulphuric acid between 3.39-12 mL. A standard volume of 100 mL was
added to the mixture and then, the tubes were placed on the Kjeldahl digestor which was
previously heated at 370ºC. Heating must be running until white fumes appeared
(digestion time near 90 minutes) or must be continue for 30 minutes more (digestion time
of 120 minutes). After that, digestion residue must be cooled and diluted up to 50 mL
with nanopure water. Distillation must be performed by adding variable volumes (25-50
mL) of distillation reagent (NaOH/Na2S2O3). Distilled ammonia must be picked up on 25
ml of 0.04 N sulphuric acid and the distillation finished when the volume was 80 mL.
Collected solutions were diluted up to 100 mL. Before standard procedure the pH of the
solutions was adjusted to 10.5.
TABLE 2. Different Kjeldahl conditions assayed.
Condition
m(K2SO4)
(g)
m(HgO)
(g)
V(H2SO4)
(mL)
Digestion
Time (min)
V(NaOH/Na2S2O3)
(mL)
1 2.224 0.0332 3.39 90 25
2 2.224 0.0332 3.39 120 25
3 10.5 0.525 12 120 50
4 6.7 0.1 10.2 120 30
5 6.7 0.1 10 120 40
6 6.7 0.525 10 120 30
Water samples
Environmental water samples with unknown ammonium concentration were
analysed. They were named as S1: Irrigation ditch water, S2: Residual water from a
factory and S3: fountain water.](https://image.slidesharecdn.com/meseguer-170216182421/85/Meseguer-251-320.jpg)
![228
Sample treatment
The samples or standards were treated in order to eliminate residual chloride and
cation interference, according to the reference standard method [15]. A 100 mL portion of
water (real or standard) was subjected to sample treatment by adding 1 mL of dechlorant
sodium sulphite 7.14 mM and 1 mL of zinc sulphate heptahydrated 0.348M. The formed
precipitate was filtered and the first 25 mL of the sample were wasted, collecting the rest
of the sample. The pH was adjusted to 2 with concentrated H2SO4 for sample
conservation. Sample pH was adjusted to 10.5 before OPA/NAC or Nessler reactions
were carried out.
OPA/NAC reaction: 1 mL of treated standards or sample for S1 and S3, and 0.1 mL
diluted up to 1 mL for S2, were analysed by adding reagents as explained above for
standards.
Nessler reaction: 2.4 mL of treated standards or sample for S1 and S3, and 0.1 mL
diluted up to 2.4 mL for S2, were analysed by adding reagents as explained above for
standards.
Kjeldahl treatment
100 mL of sample were treated by Kjeldahl method (condition 6, Table 2) for S1 and S3;
for S2, 7 mL of sample was diluted up to 100 mL before applying Kjeldahl method.
Kjeldahl method was applied in triplicate for each sample. When OPA/NAC reaction was
applied, 1 mL of digested sample for S1 and S3, and 0.3 mL of digested sample for S2
diluted up to 1 mL, were used. When Nessler method was applied, the volumes of
digested sample employed were 2.4 mL for S1 and S3, and 0.5mL diluted up to 2.4 mL
for S2.
Calculations
The data were aligned according to the maximum emission signal for Principal
Component Regression (PCR). For all calculations, Unscrambler (CAMO, Norway) and
Matlab for Windows (Math Works, Natick, MA) were used.
RESULTS AND DISCUSSION
Calibration step
According to a previous work [12], standards of Methylamine, Ethylamine, N-
Propylamine, Isopropylamine, Butylamine, Pentylamine, Hexylamine, and β-
phenylethylamine provided similar response between them when concentration was
expressed in mg/L of Nitrogen, with %Recovery respect to methylamine signal of 100,
93.5, 101.1, 93.4, 89, 100.4, 108.2 and 99 % for each amine, respectively. The same
emission maxima located at 440 nm were also obtained for all of them exciting at 333 nm
(Figure 1). The additivity between different amine signals was tested analysing mixtures
of amines (see experimental section). %Recoveries obtained were between 81-88%. With
these results, it was corroborated that the amine response in the OPA-NAC reaction was
not dependent on the primary amine, and methylamine could be used as a representative
compound.](https://image.slidesharecdn.com/meseguer-170216182421/85/Meseguer-252-320.jpg)


![231
The obtained emission spectra exciting at 415 and 333 nm for the standards
(Table 1) were processed by using principal component regression (PCR) in order to
develop a multivariate model to ammonium and amine, respectively. Top-Down
selection (TDS) and Best-Subset selection (BSS) have been employed to perform the
calibration step [16]. PCR models were obtained using as X-block the data (column
centred). The number of variables, the percentage of explained variance, the number of
factors and root mean squared errors of prediction (RMSECV) by leave-one-out cross-
validation are given in Table 4. RMSECV values were calculated in order to evaluate the
prediction ability of models. For methylamine there are no differences between the two
options of selection, and a model with complexity three has been selected. For
ammonium PC number 2 is not included in the model with BSS selection, and only PCs
number 1 and 3 have been used (complexity two). In a second step, variable selection on
the optimised model by using the Uninformative Variable Elimination (UVE) approach
has been used [17]. Only 32 and 29 original variables have been used for methylamine
and ammonium, respectively. In both cases a final model complexity two has been
selected. The RMSECV values obtained are good as can be seen in Table 4 for all
models, although UVE-BSS models were selected.
TABLE 4. Characteristics of Principal Component Regression Models obtained for
Ammonium and Methylamine by using Top-Down Selection, Best Subset Selection or
Uninformative Variable Elimination-Best Subset Selection.
Ammonium
(ëexcitation 415 nm)
Amine
(ëexcitation 333 nm)
TD BSS UVE-BSS TD BSS UVE-BSS
Number of
variables
231 231 32 180 180 29
Selected PCs 1,2 1,3 1,2 1,2,3 1,2,3 1,2
Explained
variance,
block X
99.999 99.999 99.999 99.999 99.999 99.999
rmsecv 0.089 0.051 0.052 0.034 0.037 0.040
Accuracy and precision
Taking into account previous results, the standards were analysed. The
measurements were performed exciting at 415 and 333 nm and using the ammonium
calibration graph at 485 nm and the ammonium and amine calibration equations at 440
nm, respectively. Table 5 shows the ammonium and amine concentration found in the
different mixtures assayed. Satisfactory results were obtained in all cases with relative
errors for amine and ammonium contents between 7-8 % and 4-24 % and relative
standard deviations between 6-13 % and 1-9 %, respectively.
Te same mixtures were analysed by using the PCR model. Improved results were
obtained for accuracy of ammonium. The relative errors found were between 1-16 %.](https://image.slidesharecdn.com/meseguer-170216182421/85/Meseguer-255-320.jpg)



![236
0.07), (213 ± 3) and (2.103 ± 0.018) mg/L ammonium for ditch, industrial waste and
fountain waters, respectively, which were similar to those obtained by Nessler (t test for
paired samples á > 0.05): (0.223 ± 0.018), (232 ± 3), (2.14 ± 0.02) mg/l ammonium
respectively. These results are in agreement of the water nature, for natural water with
low pollution, the N-Kjeldahl will not exceed 5 mg/l. For industrial waste water, this
parameter can be higher than 200 mg/L of ammonium. Bearing in mind the results of
Table 5, other amino compounds besides ammonium and primary aliphatic amines, are
present in these waters.
Conclusions
In this article, the use of OPA/NAC reagent for the simultaneous determination of
ammonium and primary aliphatic amines has been proposed. Bivariate calibration or
multivariate Principal Component Regression can be used obtaining similar results.
Methylamine can be used as model compound, because the response of each amine was
the same when it was expressed in mg/L of Nitrogen, and there was additivity in their
signal when mixtures were processed.
Similar calibration curves were obtained for ammonium (0.25-1.75 mg/L) with a
fixed amine concentration ranged from 0.25 to 0.625 mg/l methylamine exciting at 415
nm. No response for amines was obtained working at this wavelength. Slopes of the
Methylamine calibration curves (0.25-0.625 mg/l) were also similar with a fixed
ammonium concentration ranged from 0.25 to 1.75 mg/l exiting at 333 nm. The signal of
the ammonium affecting ordinate values.
Kjeldahl treatment was applied obtaining similar slopes for ammonium
calibration graphs before and after the treatment. Amine conversion into ammonium was
near 100% for every amine studied alone or in mixtures. The application of Kjeldahl
treatment to real water samples gave results in agreement of the water nature, and Nessler
method was applied in order to validate accuracy of the proposed method, providing
similar results to those obtained by the OPA/NAC method.
Acknowledgements
The authors are grateful to the Ministerio de Ciencia y Tecnología of Spain for
financial support received for the realization of Project PPQ 2000-1641. S.M.Ll.
expresses her gratitude to Ministerio de Educación y Cultura (Spain) for the predoctoral
grant.
References
[1] A. Aminot, D.S. Kirkwood and R. Kérouel. Mar.Chem. 56 (1997) 59.
[2] C.Dodeigne, L.Thunus, R.Lejeune. Talanta, 51 (2000) 415.
[3] D.Sahasrabuddhey, A.Jain, K.K.Verma. Analyst, 124 (1999) 1017.](https://image.slidesharecdn.com/meseguer-170216182421/85/Meseguer-260-320.jpg)
![237
[4] M. Kaminski, D.Jastrzebski, A.Przyjazny, R.Kartanowicz. J.Chromatogr. A, 947
(2002) 217.
[5] F. Dai, V.P. Burkert, H. N. Singh and W.L. Hinze. Microchem. J. 57 (1997) 166.
[6] P. Campíns-Falcó, C. Molins-Legua, A. Sevillano-Cabeza and L. A. Tortajada
Genaro, J. Chromatogr. B. 759 (2001) 285.
[7] H.Mana, U.Spohn. Fresenius J.Anal.Chem. 366 (2000) 825-829.
[8] A.Aminot, R.Kerouel, D.Birot. Wat.Res. 35 (7) (2001) 1777-1785.
[9] S.Meseguer-Lloret, C.Molins-Legua, P.Campins-Falcó. Intern.J.Envir.Anal.Chem. 82
(7) (2002) 475-489.
[10] A. Aminot, R.Kerouel, D.Birot, Wat.Res. 35-7 (2001) 1777-1785
[11] H.Mana and V.Spohn, Fresenius J Anal Chem, 366, 825-829 (2000).
[12] A.Sevillano Cabeza, Y.Moliner Martinez, C.Molins Legua, P.Campíns Falcó.
Anal.Bioanal.Chem. 376 (2003) 918-922.
[13] F. Burriel. Química analítica Cualitativa, 17th
Edn., Pub Paraninfo, Madrid (2000).
[14] Application Note ASN 52/85. Tecator Manual Digestion system 1007 (1985).
[15] S.A.Díaz-Santos. Métodos Normalizados para el análisis de aguas potables y
residuales, 17th
Edn., Apha-Awwe-WPCF, Madrid (1992).
[16] J.Verdú, D.L.Massart. Applic. Spectrosc. 52 (1998) 1425-1434.
[17] V.Centner, D.L.Massart, O.E. de Noord, S.de Jong, B.M.Vandeginste and C.Stena.
Anal.Chem. 68 (1996) 3851.](https://image.slidesharecdn.com/meseguer-170216182421/85/Meseguer-261-320.jpg)


![240
INTRODUCTION
Nitrogen is a key nutrient in natural waters, but excess N inputs lead to
eutrophication. The dominant N species in waters are: dissolved inorganic N (ammonium,
nitrite and nitrate), dissolved organic N (the largest fraction is made up of amino acids
and peptides and it is often called amino N) and particulate organic (due to small
organisms: algae, bacteria) and inorganic N [1]. Inputs of dissolved organic nitrogen
(DON) to natural waters are largely a result of autochthonous biological processes.
Additionally, external sources of DON arise from sewage and industrial effluents,
terrestrial run-off and atmospheric deposition [2]. DON content in water has largely been
ignored, difficulties in measuring DON, its chemical character remains poorly described
and an underlying assumption that it is biologically inert can be the causes [2].
Some analytical procedures exist for quantifying dissolved inorganic N (DIN), total dissolved
nitrogen (TDN) but not for dissolved organic N as mentioned above. The most used is the Kjeldahl
wet chemical method [3-6] because it is the reference method established by European Directive
[7] relative to measurement methods in the analysis of superficial waters. Kjeldahl
method was developed specifically for proteinaceous nitrogen and some authors indicated
incomplete recovery of nitrogen for some compounds [8,9]. It provides DON and
ammonium contents.
In other protocols DON is calculated as the difference between independent
measurements of TDN and DIN, but they are even more time-consuming than Kjeldahl
method. The main methods for TDN determinations are: alkaline persulfate oxidation or
UV-photo-oxidation to nitrate and oxidative combustion methods to NO [2,8].
Some papers have been published comparing several of the used methods
[2,10,11]. More work is needed in order to improve accuracy of DON results and reduce
time and/or difficulty of the technique.
Based on previous studies for ammonium [12] and the known reaction between
amine groups and hypochlorite, we propose a continuous flow system for organic amino
N and ammonium nitrogen determination on untreated water samples, in which amino
group react with hypochlorite reagent to form a chloramine and, subsequently, the
mixture reacts with luminol, generating a chemiluminescence signal. The signal emission
at 425 nm, registered as a function of time, decreases as nitrogen concentration increases,
due to a decreasement on hypochlorite concentration that depends on the amino group
present in the sample. A large number of nitrogen compounds has been assayed and
sensitivities compared, in mg/L of Nitrogen. A unique calibration graph can be used for a
great number of compounds tested and then, the method can be employed for estimating
the main dissolved organic nitrogen and ammonium. Characteristics of the method, such
as selectivity and reproducibility, have been evaluated.
The chemiluminescence method was applied to the analysis of several kind of
real water samples (natural water, lake water, irrigation ditch water, fountain water,
residual water and seawater). A comparative study of ammonium nitrogen contents
before and after Kjeldahl treatment has been carried out. Standard additions method was](https://image.slidesharecdn.com/meseguer-170216182421/85/Meseguer-264-320.jpg)
![241
applied for all samples studied in order to test the presence or absence of proportional
systematic errors.
EXPERIMENTAL
Apparatus
All spectrofluorimetric measurements were made on a Hitachi F-4500, 700v
fluorescence spectrophotometer (Tokio, Japan). The light emission was monitored at 425
nm.
The pH was measured with a Crison micropH 2000 pH-meter. A Nanopure II
(Synbron Barnstead) water unit was used.
For FI assembly (see Figure 1), a Gilson Miliplus peristaltic pump was used to
drive the reactants through the flow cell; it always worked at a flow rate of 4.1 ml/min. A
400 µL reactor was employed to perform the reaction between Hypochlorite and Nitrogen
compounds. The sample loop employed had a 50 µL internal volume. Tygon tubing (i.d.
0.8 mm) was used with the peristaltic pump. The other tubing was made of PTFE (i.d. 0.5
mm). The flow cell was a laboratory-made bundle cell [13], consisting of knotted
transparent poly-tetrafluoroethylene tube, measuring 50 cm in length and 0.8 mm of
internal diameter. The bundle cell (1.5x1x1 cm) was placed in a plastic cell. A second
kind of flow cell studied was a laboratory-made spiral cell [13] consisting of coiled
transparent PTFE tube measuring 50 cm in length and 0.8 mm of internal diameter. Its
dimensions were 1 cm (internal diameter) and 3 cm (external diameter).
For sample Kjeldahl treatment, the Digestion Unit was a Tecator Digestion
system 6, 1007 Digestor (Höganäs, Sweden), and the Distillation Unit was a Büchi 323
Distillation Unit (Switzerland).
Reagents and solutions
All solutions were prepared in nanopure water, and all reagents were of analytical
grade.
Stock standard solutions of ammonium were prepared by dissolving ammonium
chloride (Probus, Barcelona, Spain) in nanopure water.
The following amines from Sigma (Steinheim, Germany) were employed:
methylamine (MA), ethylamine (EA), n-propylamine (nPA), isopropylamine (iPA),
butylamine (BA), pentylamine (PeA), hexylamine (HA), dimethylamine (DMA),
diethylamine (DEA), trimethylamine (TMA), triethylamine (TEA), β-phenylethylamine
(BPEA), 1,7-diaminoheptane (DAH), putrescine (Put), cadaverine (Cad), spermine (Spm)
and spermidine (Spd).
The following aminoacids from Aldrich (Steinheim, Germany) were employed:
aspartic acid, tryptophan, glutamic acid, glicocole, proline, serine, tyrosine, alanine,
phenylalanine, valine, leucine, isoleucine, treonine, lisine, metionine, histidine, cysteine,
histidine and arginine.
Other solutions (1000 mg/L) were prepared: Mercaptoethanol (Scharlau,
Barcelona, Spain); Urea and sodium sulphide from Prolabo, Fontenay, France; N-Acetyl-
L-Cysteine, s-Triazine and albumine from bovine serum were from Fluka (Switzerland);](https://image.slidesharecdn.com/meseguer-170216182421/85/Meseguer-265-320.jpg)
![242
Moreover, metal solutions (0.05 M) of chromium chloride, cobalt (II) chloride, copper
(II) chloride, iron (III) chloride, cadmium chloride and mercury (II) chloride from
Panreac (Barcelona, Spain) were prepared.
The following reagents were also used: sodium hypochlorite 15%, sodium
thiosulphite and potassium sulphate from Prolabo (Fontenay, France); sodium hydroxide,
sulphuric acid and sodium carbonate from Merck (Darmstadt, Germany), and red mercury
oxide from Panreac (Barcelona, Spain).
PROCEDURES
FI procedure
The flow injection assembly is shown on Figure 1. Initial concentrations and
flow injection system assembly were the ones described in [12], with some modifications.
Firstly, hypochlorite was not electrogenerated, in order to have a simpler system.
Secondly, the sample was previously mixed with hypochlorite in a reaction coil of 400
µL in order to form the chloramine compound and, then, sample loop was filled up. A
water carrier picked up the loop contents when injection valve was turned, and it was
mixed with luminol / buffer carrier. The distance between the last T-junction and the
detection cell was the minimum possible, 5 cm.
The working solutions were as follows: hypochlorite 0.9 mM (in
hydrogencarbonate / carbonate buffer 0.144 mM, pH 11.2), and luminol 1 mM (in
hydrogencarbonate / carbonate buffer 0.2 M, pH 11.2).
FIGURE 1. Flow injection assembly for chemiluminescence organic nitrogen determination.
The following standard solutions were prepared and measured according to the
methodology proposed:
Standard solutions of ammonium, amines, amino acids, N-acetyl-L-cysteine
(NAC), s-triazine and urea were measured in the range of 0-4 mg/L of nitrogen.
Albumine from bovine serum was measured in the range of 2.5-100 mg/L of albumine.
W
Detection
cell
Sample
loop: 50 µL
IV
Reactor,
400 µL
Standard/Sample
H2O
Luminol 1 mM,
0.2M CO3
2-
/HCO3
-
pH 11.2
Hypochloride 9 µM,
0.144mM CO3
2-
/HCO3
-
pH 11.2
v = 35 rpm
W](https://image.slidesharecdn.com/meseguer-170216182421/85/Meseguer-266-320.jpg)
![243
Standards of cysteine, metionine, NAC, mercaptoethanol and sodium sulphide
were prepared in the range of 0-5.4· 10-5
M of sulphide.
Metal solutions of: Copper (II) (2· 10-6
, 4· 10-5
and 5· 10-4
M), Fe(III) (9· 10-6
, 5· 10-4
and 5· 10-3
M), Cd(II) (4.5· 10-8
, 2.5· 10-4
and 2.5· 10-3
M), Hg(II) (5· 10-9
, 6.25· 10-3
and
0.05M), Cr(III) (1· 10-5
M) and Co(II) (8.5· 10-6
M) were also measured.
Kjeldahl treatment
For the application of Kjeldahl treatment [14], 6.7 g of potassium sulphate, 0.52 g
of red mercury oxide and 10 mL of concentrated sulphuric acid, were added to 100 mL of
nitrogen containing compound standard in a digestion tube. Then, the tubes were placed
on the Kjeldahl digestor, previously heated at 370ºC. Heating is kept until white fumes
appeared and, then, digestion continues for 30 minutes more. After that, digestion residue
must be cooled and diluted up to 100 mL with nanopure water. Distillation is performed
after the addition of 35 mL of distillation reagent (25 g of sodium thiosulphate and 500 g
of sodium hydroxide diluted up to 1L with nanopure water). Distilled ammonia must be
picked up on 25 mL of 0.04 N sulphuric acid until 80 mL. This solution was diluted up to
100 mL. Before experimental measures, collected solutions were adjusted to pH 6.0-7.0.
Solutions (100 mL) of TEA, urea, histidine and arginine, with final
concentrations of 1.5 mg/L of nitrogen, solution mixtures of MA / PeA / DMA or MA /
PeA / DMA / TEA, with final Nitrogen concentration of 1.5 mg/L or 1.8 mg/L
respectively, and a solution with 1 g/L of albumine, were submitted to Kjeldahl treatment.
Sample procedures
Eight different water samples were collected and pH adjusted to 2.0 with
concentrated nitric acid until they were used. At the moment of the analysis, the samples
pH value was adjusted near 7.0. All of them were analysed directly by the proposed FI
chemiluminescence method, before and after the Kjeldahl treatment. Samples were
named as follows: S1: mineral bottled water; S2: tap water; S3 and S7: irrigation ditch
water (from different sources); S4: lake water; S5: sea water; S6: industrial waste water
and S8: fountain water with clear aspect of eutrophication.
4.45 mL diluted up to 5 mL for S1, S2, S3, S4, S7 and S8, 1ml diluted up to 5 mL
for S5, or 0.035 mL diluted up to 5 mL for S6, were prepared and measured in triplicate.
These samples were also spiked with ammonium (0.75, 1.5 and 3 mg/L of nitrogen), in
order to apply the standard additions method.
Kjeldahl treatment
Kjeldahl treatment was applied, as described above for standards, to 100 mL of
S3, S4, S5 and S8 (all in triplicate), and to 7 mL of S6 diluted up to 100 mL (in
triplicate). After Kjeldahl treatment, 4.45 mL diluted up to 5 mL for S3, S4, S5 and S8, or
0.35 mL diluted up to 5 mL for S6, were measured in triplicate. They were also spiked
with ammonium (0.75, 1.5 and 3 mg/L of nitrogen), to apply the standard additions
method.](https://image.slidesharecdn.com/meseguer-170216182421/85/Meseguer-267-320.jpg)




![248
their structure were contributing to the decrease in chemiluminescence. As can be
demonstrated below this atom was S.
TABLE 4. Percentage of recovery (slopeaminoacid*100/slopeNH4+) found for aminoacids
individually and mean %recovery.
AMINOACID
% Recovery
bamine*100/bNH4+
Mean Recovery
(% REC ± s)
aspartic acid 93.5
tryptophan 87.1
glutamic acid 101.9
glicocole 97.6
proline 96.6
serine 99.9
tyrosine 95.4
almandine 98.1
phenylalanine 97.1
valine 100.6
leucine 94.3
isoleucine 96.0
treonine 97.1
lysine 93.8
(96 ± 4)%
Selectivity of the method: Interference study
The first type of interfering compounds studied were those containing sulphur
atoms, because they can produce interferences in the nitrogen determination as can be
seen in the previous section for amino acids containing S. Two compounds, sodium
sulphide (S2-
) and mercaptoethanol (R-SH), without nitrogen content, were studied. As it
was expected, both compounds lowered the chemiluminiscence signal. Sulphur
interference was twice the mercaptoacetic interference, the slopes of the calibration
graphs obtained were 3058 and 1850 L/mg S.
The response for cysteine, amino acid with sulphur content, and its derivative N-
acetyl-cysteine were due to the contribution of the both atoms, N and S. The experimental
signals were around 100 % of the theoretical signals obtained bearing in mind the slopes
of the calibration graphs of ammonium and mercaptoethanol. The interference caused by
the R-S-R group is small as can be derived from the recovery given in the previous
section for metionine. The concentration of biogenic thiols in natural waters have been
found to be about 0.25 ppb [15], and the higher limit admitted for drinking water for H2S
is 50 ppb [7], at these levels the sulphur containing compounds did not interfere in the
reaction, giving the same signal that the corresponding to the blank solution.](https://image.slidesharecdn.com/meseguer-170216182421/85/Meseguer-272-320.jpg)


![251
after the Kjeldahl digestion (this is the case of S6 and S7). S6 was also analysed by the
Nessler and o-phthaldialdehyde/N-Acetyl-L-Cysteine (OPA/NAC) methods for
ammonium [16], and after Kjeldahl treatment, the found concentrations were (180 ± 3,
n=3) and (166 ± 3, n=3) mg/L N for Nessler and OPA/NAC procedures respectively. For
this type of samples, we could replace the tedious Kjeldahl treatment by the application of
the direct simple FI system presented in this work. The second group of samples, showed
different nitrogen concentrations before and after the Kjeldahl digestion (this is the case
of S4, S5 and S8). The proposed method for S4 provided 60 % of the N-Kjeldahl, S5
presented increase of the blank signal and then can not be analysed by the FI method
directly as mentioned previously. The results for S8 can be explained by the presence of
nitrogen containing compounds that does not react completely with the hypochlorite, but
are transformed into ammonium after Kjeldahl treatment or, more probably, by the
presence of complex organic matrix, decomposed by the acidic digestion (in a similar
way that albumine results predict). S8 was also analysed by the Nessler and OPA-NAC
method for ammonium [16], the results were (0.25±0.02, n=3) and (0.29±0.02, n=3)
mg/L N, respectively before Kjeldahl treatment, and (1.66 ± 0.02, n=3) and (1.635 ± 0.18,
n=3) mg/L N, respectively after Kjeldahl treatment. These results are near those obtained
with the proposed chemiluminescence FI detection procedure.
TABLE 7. Total Nitrogen content (Organic and ammonium)(mg/L N) found in different water
samples studied, before and after Kjeldahl treatment. Samples analysed in triplicate.
Nitrogen Content (mg/L N)
Sample
Before Kjeldahl Treatment After Kjeldahl Treatment
S4 (0.92 ± 0.15) (1.59 ± 0.06)
S5 N.D. (1.66 ± 0.10)
S6 (140 ± 8) (149 ± 11)
S7 (0.52 ± 0.04) (0.57 ± 0.03)
S8 (0.35 ± 0.05) (1.99 ± 0.07)
CONCLUSIONS
The response of amino compounds (-CH2-NH-) including ammonium by
applying the proposed methodology gave recoveries near 100% for primary and
secondary aliphatic amines, polyamines, arylalkylamines and fourteen of the aminoacids
assayed. Tertiary amines and aromatic or resonant nitrogens did not give the reaction, N-
proteins is incompletely recovered. The reaction was interfered by sulphide groups, but
not at concentrations below their legislation level. Metal ions studied have not resulted
interferences below their legislation levels. The method is capable of estimating total
dissolved N-amino in a sample very quickly.
Matrix effects have not been observed in fortified samples, and accurate results
have been obtained with recoveries near 100%. Results shown in this paper permit to
establish that for some kind of samples, Kjeldahl treatment can be omitted and the
simpler FI system presented in this work used. This option is very advantageous because](https://image.slidesharecdn.com/meseguer-170216182421/85/Meseguer-275-320.jpg)
![252
the method does not require sample treatment, three replicates of the sample can be made
in 1 min, toxicity of the reagents is low and the method can be used in in situ monitoring .
The proposed methodology can be also used as an alternative for final ammonium
detection after Kjeldahl digestion of the samples. The precision of the method for
ammonium determination was very good, with RSD of 0.3-3.7 % for intraday
repeatibility values, and 1.9% for interday reproducibility of the slope calibration line
values. These values were similar to Nessler method precision (%RSD=0.4%), and better
than OPA-NAC method precision (%RSD below 12.5%). Dynamic range was 0.24-4
mg/L N. For Nessler method, it was 0.66-3.88 mg/L N and for OPA-NAC method it was
0.155-1.088 mg/L N. Detection limit reached with the proposed method (0.07 mg/L N)
was similar to that obtained with OPA-NAC method (0.054 mg/L N), and better than that
reached with Nessler reference method (0.46 mg/L N). The rapidity of the proposed
method resulted excellent (60 samples/hour) compared with Nessler (6 samples/hour) or
OPA/NAC (10 samples/hour) methods.
ACKNOWLEDGEMENTS
The authors are grateful to the Ministerio de Ciencia y Tecnología of Spain for
financial support received for the realization of Project BQU2003-06138 . Susana
Meseguer Lloret expresses her gratitude to Ministerio de Educación y Cultura (Spain) for
the predoctoral grant.
BIBLIOGRAPHY
[1] T.P. Burt, A.L. Heathwaite, S.T. Trudgill. Nitrate: Processes, Patterns and
Management. John Wiley & Sons, New York 1993.
[2] E.-S. A. Badr, E.P. Achterberg, A.L. Tappin, S.J. Hill, C.B. Braungardt, Trends Anal.
Chem. 22 (2003) 819.
[3] K.Cameron, C.Madramootoo, A.Crolla, C.Kinsley. Wat. Res. 37 (2003) 2803.
[4] S.E.Tsiouris, A.P.Mamolos, K.L.Kalburtji, D.Alifrangis,Agric. Ecosyst. & Envir., 89
(2002) 117.
[5] S.H. Ensign, M.A. Mallin, Wat. Res., 35 (2001) 3381.
[6] V. P. Aneja, B. Bunton, J. T. Walker, B. P Malik, Atm. Environ., 35 (2001) 1949.
[7] Directive 98/83/CEE.
[8] B.M. Jones, C.G. Daughton, Anal. Chem. 57 (1985) 2320.
[9] EPA-821-R-01-004. Method 1687. 2001
[10] G. Alavoine, B. Nicolardot, Analusis 28 (2000) 88.
[11] J. H. Sharp et al., Marine Chemistry 78 (2002) 171.
[12] J. Li, P. Purnendu, K. Dasgupta, Anal. Chim. Acta 398 (1999) 33.
[13] P.Campins-Falcó, L.A.Tortajada-Genaro, F.Bosch-Reig, Talanta. 55 (2001) 403-
413.
[14] Application Note ASN 52/85. Tecator Manual Digestion system 1007 (1985).
[15] D. Tang, L.S. Wen, P.H. Santschi, Anal. Chim. Acta, 408 (1-2) (2000) 299-307.
[16] S.Meseguer-Lloret, C.Molins-Legua, P.Campíns-Falcó, Intern. J. of Environ. Anal.
Chem. 82 (7) (2002) 475-489.](https://image.slidesharecdn.com/meseguer-170216182421/85/Meseguer-276-320.jpg)
Meseguer
- 1. DEPARTAMENTO DE QUÍMICA ANALÍTICA MÉTODOS QUIMIOLUMINISCENTES EN QUÍMICA ANALÍTICA SUSANA MESEGUER LLORET UNIVERSITAT DE VALENCIA Servei de Publicacions 2004
- 2. Aquesta Tesi Doctoral va ser presentada a Valencia el día 01 de Juliol de 2004 davant un tribunal format per: - D. Francisco Bosch Reig - Dª. Mª Dolores Pérez Bendito - D. Ángel Ríos Castro - D. Miguel Valcárcel Cases - Dª. Adela Sevillano Cabeza Va ser dirigida per: Dª. Pilar Campíns Falcó Dª. Carmen Molins Legua D. Jorge Verdú Andrés ©Copyright: Servei de Publicacions Susana Meseguer Lloret Depòsit legal: I.S.B.N.:84-370-6001-X Edita: Universitat de València Servei de Publicacions C/ Artes Gráficas, 13 bajo 46010 València Spain Telèfon: 963864115
- 3. I UNIVERSITAT DE VALÈNCIA FACULTAD DE QUÍMICA DEPARTAMENTO DE QUÍMICA ANALÍTICA MÉTODOS QUIMIOLUMINISCENTES EN QUÍMICA ANALÍTICA TESIS DOCTORAL SUSANA MESEGUER LLORET Abril 2004
- 4. II
- 5. III UNIVERSITAT DE VALÈNCIA FACULTAD DE QUÍMICA DEPARTAMENTO DE QUÍMICA ANALÍTICA La Dra. Dña. Pilar Campíns Falcó, Catedrática del Departamento de Química Analítica de la Facultad de Ciencias Químicas de la Universidad de Valencia, la Dra. Dña. Carmen Molins Legua, Titular del Departamento de Química Analítica de la Facultad de Ciencias Químicas de la Universidad de Valencia, y el Dr. D. Jorge Verdú Andrés, Titular del Departamento de Química Analítica de la Facultad de Ciencias Químicas de la Universidad de Valencia, CERTIFICAN: Que la presente Memoria, "Métodos quimioluminiscentes en Química Analítica", realizada en el Departamento de Química Analítica de la Universidad de Valencia, constituye la Tesis Doctoral de Dña. Susana Meseguer Lloret. Así mismo, certifican haber dirigido y supervisado tanto los diferentes aspectos del trabajo como su redacción. Y para que conste a los efectos oportunos, firmamos la presente en Valencia a 1 de Abril de 2004. Dra. Pilar Campíns Falcó Dra. Carmen Molins Legua Dr. Jorge Verdú Andrés
- 6. IV
- 8. VI
- 9. VII AGRAÏMENTS • A Pilar, a Carmen i a Jorge, per tots els coneixements transmesos durant aquestos anys, tant a nivell professional com personal. Gràcies també per l’amistat brindada, això restarà per sempre. • Als meus pares, a les meues germanes, cunyats i a tota la meua família, per animar- me a fer açò i per estar sempre al meu costat. Sense vosaltres, no hauria arribat fins ací. • A Andrés, per viure amb mí cada moment d’esta Tesis, per saber entendre’m en els moments durs i per haver compartit amb mi els moments de felicitat. • A Eli i a Marc per el somriure que em lliuren cada dia. • A Rosa i a Adela, per estar ahí quan vos he necessitat. • A Yolanda, a Xelo, a Luis i a Alberto, per els dinars compartits, els moments de recolçament al laboratori quan creus que les coses no t’eixiran mai, i també per les rises que hem compartit i l’amistat que hem creat. • A Miguel Valcárcel, a Marisol Cárdenas, a Mercedes Gallego i a tots els companys del seu grup d’investigació, per haver-me acollit durant tres mesos i haver-me donat l’oportunitat de treballar amb ells. • A Ángeles i a Marta perquè els anys no han deixat anar la nostra amistat. • A les meues amigues de la Facultat, per estar sempre tan pendents, per els cinc anys de carrera compartits, i per els que ens queden per viure juntes. • Als meus amics i amigues per el recolçament demostrat.
- 10. VIII
- 11. IX ÍNDICE TÍTULO ...................................................................................................................... I INDICE ....................................................................................................................... IX LISTADO DE FIGURAS .......................................................................................... XV LISTADO DE TABLAS ............................................................................................ XVIII LISTADO DE SÍMBOLOS Y ABREVIATURAS .................................................. XXI OBJETIVOS............................................................................................................... 1 1. INTRODUCCIÓN .................................................................................................. 7 1.1. Consideraciones sobre el análisis de agua y analitos estudiados ................... 9 1.1.1. Características de las aguas ................................................................... 9 1.1.2. Características de los analitos estudiados y legislación aplicable a los mismos ............................................................................................................. 12 1.1.2.1. Descripción de los analitos estudiados .................................. 12 1.1.2.2. Legislación aplicable ............................................................. 13 1.2. Características y tende ncias del análisis medioambiental ............................ 15 1.3. Quimioluminiscencia ........................................................................................... 18 1.3.1. Perspectiva Histórica ............................................................................. 18 1.3.2. Fundamentos y Mecanismos ................................................................. 20 1.3.3. Características de la quimioluminiscencia como técnica analítica ....... 21 1.3.3.1. Ventajas y limitaciones de la técnica ..................................... 21 1.3.3.2. Tendencia actual .................................................................... 23 1.3.4. Reacciones Quimioluminiscentes .......................................................... 24 1.3.5. Reacciones quimioluminiscentes directas en fase líquida ..................... 26 1.3.5.1. Reacciones del Luminol y sus análogos ................................ 26 1.3.5.2. Reacción de los ésteres de acridinio ...................................... 28 1.3.5.3. Reacción de Tris (2,2’-bipiridina) rutenio (III) ..................... 29 1.3.5.4. Reacciones bioluminiscentes ................................................. 29 1.3.5.5. Reacciones de oxidación directa ........................................... 30 1.3.6. Reacciones quimioluminiscentes indirectas en fase líquida .................. 30 1.3.6.1. Reacciones con dioxoetanos (directas e indirectas) .............. 30 1.3.6.2. Reacciones con peroxioxalatos .............................................. 31 1.4. Reactivos .............................................................................................................. 34 1.5. Aparatos, montajes, instrumentación y software ............................................. 36 1.6. Bibliografía .......................................................................................................... 42
- 12. X 2. RESULTADOS OBTENIDOS Y DISCUSIÓN ................................................... 47 2.1. REACCIÓN DEL TCPO .................................................................................... 49 2.1.1. Screening de aminas alifáticas y disminución de sus límites de detección en aguas ......................................................................................... 49 2.1.1.1. Preconcentración y dansilación de aminas alifáticas utilizando cartuchos C18 de extracción en fase sólida. Aplicación al análisis de screening en muestras medioambientales de agua ............ 49 Derivatización en disolución ................................................. 52 Preconcentración y derivatización en cartuchos C18 ............ 52 Separación cromatográfica .................................................... 55 Características analíticas ........................................................ 55 Análisis de muestras de agua ................................................. 58 Conclusiones ......................................................................... 60 2.1.1.2. Derivatización en línea de aminas alifáticas con Dns-Cl acoplada a HPLC con detección fluorescente .................................... 62 Condiciones de enriquecimiento, lavado y derivatización .... 62 Condiciones cromatográficas ................................................ 63 Características analíticas ........................................................ 63 Análisis de muestras de agua ................................................ 65 Conclusiones ......................................................................... 65 2.1.1.3. Determinación de aminas alifáticas en agua mediante HPLC con detección quimioluminiscente ..................................................... 67 Condiciones de preconcentración y condiciones pre y post columna ................................................................................. 68 Condiciones cromatográficas ................................................ 70 Curvas de calibrado y parámetros analíticos ......................... 71 Análisis de muestras de agua ................................................. 73 Comparación con otros sistemas de detección ...................... 75 Conclusiones .......................................................................... 76 2.1.2. Disminución de l límite de detección de amonio en agua e incremento de la exactitud de su de terminación ....................................... 77 2.1.2.1. Determinación de amonio en muestras de agua utilizando el reactivo OPA-NAC. Estudio comparativo con los métodos de Nessler y Electrodo Selectivo de amonio ........................................... 79 Método de OPA-NAC ........................................................... 79 Estudios de confirmación: Método de OPA-NAC, Nessler y Electrodo Selectivo ................................................................ 82 Revisión del Método de Nessler ............................... 82 Revisión del Método de Electrodo Selectivo ............ 83 Análisis de muestras de agua ................................................. 84 Aplicación del Método de Nessler ............................ 84 Aplicación del Método de Electrodo Selectivo ........ 85 Aplicación del Método de OPA-NAC ...................... 86 Comparación de resultados .................................................... 87 Conclusiones .......................................................................... 88
- 13. XI 2.1.2.2. Determinación quimioluminiscente de amonio en aguas por HPLC .................................................................................................. 89 Optimización de la dansilación precolumna de amonio ........ 90 Características analíticas ........................................................ 93 Análisis de muestras de agua ................................................. 95 Conclusiones .......................................................................... 99 2.1.3. Método de screening de muestras para Cu(II) ................................. 100 2.1.3.1. Método automático de inyección en flujo para el screening de muestras de agua para Cu(II), basado en la reacción quimioluminiscente de coproporfirina I-Cu (II) / TCPO / H2O2 ........ 101 Estudio de la señal del Detector de Quimioluminiscencia FireFly ................................................................................... 101 Optimización del sistema de flujo ......................................... 102 Sensibilidad y selectividad del método ................................. 103 Fiabilidad del método para el screening de muestras de agua para Cu(II) ............................................................................. 105 Aplicación a muestras de agua .............................................. 106 Conclusiones .......................................................................... 107 2.2. REACCIÓN DEL LUMINOL ........................................................................... 109 2.2.1. Mejora de la exactitud de la determinación de Cr(III), Cr(VI), y Co(II) .............................................................................................................. 109 2.2.1.1. Elaboración de una guía para la eliminación de errores sistemáticos en la especiación de Cr(III) y Cr(VI) empleando la reacción del Luminol con H2O2. Aplicación a muestras de agua ....... 110 Modelos de Calibración para Cr(III), Cr(VI) y Cr Total ....... 111 Estudio de interferencias ....................................................... 114 Aplicación a muestras reales ................................................. 115 Método de adición estándar (MOSA) ....................... 115 Muestra de HCl ......................................................... 116 Muestras de agua ...................................................... 117 Conclusiones .......................................................................... 118
- 14. XII 2.2.1.2. Influencia de los protocolos de acondicionamiento de muestras de agua en la detección quimioluminiscente de elementos traza .................................................................................................... 119 Señal QL de muestras no acidificadas (sin acondicionar) ..... 121 Señal QL de muestras acondicionadas con HCl 5· 10-3 M ..... 124 Señal QL de muestras acondicionadas con HNO3 0.5 M ...... 125 Aplicabilidad ......................................................................... 127 Conclusiones .......................................................................... 129 2.2.1.3. Calibración multivariada aplicada a la determinación quimioluminiscente simultánea de Cobalto y Cromo ........................ 130 Estudio de la señal quimioluminiscente ................................ 130 Evaluación de los modelos de calibración ............................. 131 Respuesta lineal ........................................................ 132 Respuesta no lineal ................................................... 133 Validación .............................................................................. 135 Conclusiones .......................................................................... 136 2.2.2. Determinación de Nitrógeno orgánico amino y amoniacal ............ 138 2.2.2.1. Método fluorimétrico de Roth modificado para muestras de agua: determinación simultánea de amonio y grupos amino primario y su utilidad en la determinación del Nitrógeno Kjeldahl Total ........ 139 Etapa de calibración ............................................................... 140 Precisión y exactitud .............................................................. 143 Método Kjeldahl .................................................................... 144 Análisis de muestras de agua ................................................. 148 Conclusiones ......................................................................... 149 2.2.2.2. Método quimioluminiscente automático para la estimación total del Nitrógeno orgánico (-CH2-NH-) y amonio ........................... 150 Parámetros analíticos ............................................................. 150 Respuesta de compuestos de Nitrógeno ................................ 152 Selectividad del método: Estudio de interferencias ............... 155 Tratamiento Kjeldahl ............................................................. 156 Análisis de muestras de agua ................................................. 156 Conclusiones ......................................................................... 158 2.3. REFERENCIAS BIBLIOGRÁFICAS .............................................................. 161 3. CONCLUSIONES FINALES ............................................................................... 167
- 15. XIII 4. APÉNDICES ........................................................................................................... 177 Apéndice 1. *Preconcentration and dansylation of aliphatic amines using C18 solid-packings. Application to the screening analysis in environmental water samples. 181 Apéndice 2. *Derivatization on solid supports: An alternative method for solution derivatization of amines in several matrices. 183 Apéndice 3. *Sensitive determination of aliphatic amines in water by High- Performance Liquid Chromatography with chemiluminescence detection. 185 Apéndice 4. *Ammonium determination in water samples by using OPA-NAC reagent: A comparative study with Nessler and Ammonium Selective Electrode methods. 187 Apéndice 5. Selective chemiluminescence determination of ammonium in water by HPLC. 189 Apéndice 6. FI automatic method for the determination of copper(II) based on coproporphyrin I - Cu(II) / TCPO / H2O2 chemiluminescence reaction for the screening of waters. 205 Apéndice 7. *A guide to avoid method bias of Chromium (III, VI) chemiluminescence determination by luminol-hydrogen peroxide reaction. Application to water samples. 217 Apéndice 8. *Influence of water sample storage protocols in chemiluminescence detection of trace elements. 219 Apéndice 9. *Multivariate calibration applied to simultaneous determination of cobalt and chromium. 221 Apéndice 10. Modified Roth’s fluorimetric method for water samples: ammonium and primary amine groups determination and utility in total nitrogen Kjeldahl. 223 Apéndice 11. Chemiluminescence authomatic method for total estimation of organic nitrogen (-CH2-NH-) and ammonium. 239 * De los Apéndices marcados solo se detalla la referencia bibliográfica y el enlace URL en el caso de que la versión electrónica esté contratada por la Universitat de València, ya que se trata de artículos publicados en revistas comerciales.
- 16. XIV
- 17. XV LISTADO DE FIGURAS Figura 1. Mecanismos de las reacciones quimioluminiscentes ......................................... 20 Figura 2. Intensidad de emisión quimioluminiscente frente al Tiempo ............................ 22 Figura 3. Número de publicaciones en revistas científicas de ámbito internacional frente a cada año desde 1979 a 2003 .................................................................................. 23 Figura 4. Mecanismo propuesto para la reacción del luminol ........................................... 26 Figura 5. Mecanismo de la reacción QL de los ésteres de acridinio ................................. 28 Figura 6. Mecanismo de la reacción QL de dioxoetanos .................................................. 31 Figura 7. Mecanismo propuesto para la reacción QL con peroxioxalato .......................... 31 Figura 8. A) Equipo cromatográfico HP1100. B)Detectores UV/vis y Fluorímetro ........ 36 Figura 9. Espectrofotómetro HP 8452 conectado a un PC Hewlett Packard Vector XM 5/90 ..................................................................................................................................... 37 Figura 10. Equipo cromatográfico 1050, Sistema post-columna PCX-5100, detector Jasco CL-1525 e interfaz Hercule Lite ............................................................................... 37 Figura 11. Fluorímetro modelo F4500 .............................................................................. 38 Figura 12. Fluorímetro Jasco FP-750 ................................................................................ 38 Figura 13. Detector FireFly CL ......................................................................................... 38 Figura 14. Esquema correspondiente a la derivatización en línea acoplada a la separación cromatográfica (Apéndice 2) ...................................................................... 39 Figura 15. Esquema correspondiente a la separación cromatográfica de aminas alifáticas (Apéndice 3) / amonio (Apéndice 5) acoplada a un sistema de reacción post-columna con detección quimioluminiscente ....................................................... 39 Figura 16. Sistema FIA utilizado en el Apéndice 6 .................................................... 40 Figura 17. Sistema FIA utilizado en los Apéndices 7 y 8 ........................................... 40 Figura 18. Sistema FIA utilizado en el Apéndice 11 .................................................. 41 Figura 19. Área de Pico frente a: A) pH de la reacción. B) Tiempo de reacción. C) Temperatura de la reacción. (♦ Metilamina, Etilamina, • N-Propilamina, ×× Butilamina, ∗ Pentilamina. Condiciones: DnsCl 5 mM; Tampón borato 20 mM; concentración de amina 5 mg/l.) ......................................................................................... 53 Figura 20. Cromatogramas para blanco ( ) y mezcla de aminas ( ) con detección A)UV a 333 nm, y B) Fluorescencia λexc = 350 nm / λem = 530 nm .................. 56 Figura 21. Porcentajes de recuperación en dos muestras de agua diferentes, para las diferentes aminas a diferentes niveles de concentración .................................................... 58 Figura 22. Cromatogramas para: a) Muestra 4. b) Muestra 4 fortificada con la mezcla de 1.25 mg/L de cada amina ............................................................................................... 59 Figura 23. Cromatogramas para la derivatización en línea de aminas alifáticas con Cloruro de dansilo para un blanco y una mezcla de aminas ............................................... 64 Figura 24. Cromatograma de screening de una muestra de agua residual ........................ 65 Figura 25. Porcentajes de recuperación para las diferentes aminas estudiadas en función del volumen de muestra procesado ..................................................................................... 69 Figura 26. Cromatogramas correspondientes a blanco, patrón de aminas alifáticas y patrón de aminas alifáticas + aminas biogénicas, utilizando el gradiente de Fase Móvil 1. R-reactivo, 1-MA (concentración 0.15 mg/l), 2- EA (0.240 mg/l), 3-BA (0.3 mg/l), 4- DEA (0.48 mg/l), 5-PeA (0.3 mg/l), 6-HA (0.3 mg/l), 7-Put (0.3 mg/l), 8-Cad (0.3 mg/l), 9-Spd (0.3 mg/l), 10-Spm (1.12 mg/l) ..................................................................... 70
- 18. XVI Figura 27. Cromatogramas correspondientes a blanco y patrón de aminas alifáticas + aminas biogénicas, utilizando el gradiente de Fase Móvil 2. R-reactivo, 1-MA (0.15 mg/l), 2-EA (0.240 mg/l), 3-BA (0.3 mg/l), 4-DEA (0.48 mg/l), 5-PeA (0.3 mg/l), 6- HA(0.3 mg/l), 7-Put (0.3 mg/l), 8-Cad (0.3 mg/l), 9-Spd (0.3 mg/l), 10-Spm (1.12 mg/l). 71 Figura 28. Cromatogramas correspondientes a una muestra de agua residual utilizando la Fase Móvil 1 y la fase Móvil 2. R:reagent, 1-MA, 5-PeA y 7-Put ................................. 75 Figura 29. Señal fluorescente frente al tiempo. a) λexc = 415 nm / λemisión = 485 nm; b) λexc = 333 nm / λem = 462.5 nm .......................................................................................... 80 Figura 30. Espectros de emisión de fluorescencia. a) λexc = 415 nm / λemisión = 485 nm (t reacción 5 min); b) λexc = 333 nm / λem = 462.5 nm (t reacción 2 min) ............................. 80 Figura 31. Curvas de calibrado de: 1-Amonio. 2-Amonio en presencia de MA (1 mg/l). 83 Figura 32. Curvas de calibrado de: a) Aminas. b) Aminas en presencia de 1 mg/l de amonio. (♦ metilamina, etilamina, isopropilamina, ×× β-feniletilamina) ..................... 83 Figura 33. Pendientes de las curvas de calibrado al aplicar el Método de Nessler en las diferentes condiciones estudiadas. (♦ Agua de riego (S1), Agua residual (S2), Agua de fuente (S3). Condiciones: NR1 or NR2 (Reactivo de Nessler 1 o 2); T: Muestra Tratada; NT: Muestra no tratada; TNC: Muestra tratada sin declorar; V:Volumen de muestra (mL); Volumen total=2.5 mL) .............................................................................. 84 Figura 34. Gráfico de Youden a diferentes tiempos para la muestra de agua residual ..... 85 Figura 35. Cromatogramas del blanco y patrón de amonio de 0.187 mg/L en las condiciones óptimas de derivatización ............................................................................... 91 Figura 36. Altura de pico del Blanco frente a Volumen de Acetonitrilo (Concentración de DnsCl fija 0.45 mM) o frente a concentración de DnsCl (Volumen de acetonitrilo fijo 0.4 mL) ............................................................................................................................... 92 Figura 37. Cromatogramas correspondientes a: 1. Blanco. 2. Patrón de amonio de 8µg/L (LD). 3. Mezcla de varias aminas contaminadas de amonio: MA 0.1 mg/L, EA 0.2 mg/L, N-PrA 0.3 mg/L, DMA 0.3 mg/L, BA 0.3 mg/L, DEA 0.5 mg/L, PeA 0.3 mg/L y HA 0.3 mg/L .......................................................................................................... 94 Figura 38. Cromatogramas de las muestras de agua (3 réplicas). A) Agua residual. B) Agua de lago ....................................................................................................................... 95 Figura 39. Cromatogramas de la muestra de agua de fuente fortificada con 0, 0.15 y 0.3 mg/L de amonio .................................................................................................................. 97 Figura 40. A) Espectro de emisión QL del blanco de Coproporfirina I. B) Curva cinética de la reacción QL Coproporfirina I/TCPO/H2O2 .................................................. 102 Figura 41. Registros de inyección en flujo de los patrones de la curva de calibrado de Cu(II) .................................................................................................................................. 104 Figura 42. Porcentajes de falsos positivos (%FP) y falsos negativos (%FN) en patrones de Cu(II) a concentraciones cercanas al límite de detección (DL) ..................................... 106 Figura 43. Registros de señal para la curva de calibrado de Cr(VI) con HCl 5· 10 -3 M ..... 112 Figura 44. Curvas de calibrado potenciales para Cr(VI)(♦HCl 1· 10 -3 M, HCl 5· 10 -3 M) y para Mezclas (Cr(III)+Cr(VI))( HCl 1· 10 -3 M, ×× HCl 5· 10 -3 M) ........................... 113 Figura 45. Influencia del efecto matriz en las curvas potenciales (♦Conversión 100%. Conversión 80 %. Conversión 120%.) ...................................................................... 115 Figura 46. Señales quimioluminiscentes para Cr, Co y Material Estándar de Referencia (SRM © 1640). Procedimiento en estático ......................................................................... 126
- 19. XVII Figura 47. Pendiente de la curva de calibrado en el intervalo lineal en las diferentes condiciones experimentales. 1: Sin acondicionar; 2: HCl 5· 10 -3 ; 3: HNO3 0.5; 4: EDTA 10 -2 ; 5: EDTA 1· 10 -2 , HCl 5· 10 -3 ; 6: EDTA 5· 10 -3 ; 7: EDTA 5· 10 -3 , HCl 5· 10 -3 ; 8: EDTA 10 -2 (portador KOH); 9: EDTA 10 -2 (portador tamón); 10: EDTA 5· 10 -3 (portador KOH); 11: EDTA 5· 10 -3 (portador tampón); 12: EDTA 10 -2 (portador tampón), HCl 5· 10 -3 ; 13: EDTA 5· 10 -3 (portador KOH), HCl 5· 10 -3 ; 14: EDTA 5· 10 -3 (portador tampón), HCl 5· 10 -3 . (Concentración: mol/L) .................................................... 127 Figura 48. Porcentajes de recuperación de la señal para diferentes mezclas de metales .. 128 Figura 49. Porcentajes de recuperación del SRM © 1640 respecto a mezcla de patrones en diferentes condiciones experimentales .......................................................................... 129 Figura 50. Perfiles normalizados Intensidad-Tiempo para Cr y Co .................................. 131 Figura 51. Perfiles experimentales Pendiente-Tiempo para Cr y Co, y perfil teórico de la mezcla incluyendo el error heterocedástico .................................................................... 132 Figura 52. A) Diseño experimental 5 2 . B) Gráfico de puntuaciones con las dos primeras CPs ...................................................................................................................................... 134 Figura 53. Espectros de emisión de fluorescencia para patrón de NH4 + de 1.75 mg/L (a λexc =333 nm y a λexc=415 nm) y patrón de MA de 0.375 mg/L (a λexc=333 nm y a λexc=415 nm) ....................................................................................................................... 141 Figura 54. %Recuperación de NH4 + calculado respecto a la señal (o pendiente) obtenida sin Kjeldahl, a diferentes condiciones Kjeldahl (Tabla 67). Condición 2: n=7 : 5 patrones de 0.5 mg/L de NH4 + y 2curvas de calibrado (0, 0.5, 1.25, 2, 2.75 mg/L NH4 + y 0, 0.2, 0.5, 0.9, 1.3 mg/L NH4 + respectivamente). Condición 3: n=4: 2 patrones de 0.5 mg/L de NH4 + y 2 curvas de calibrado (0, 0.05, 0.25, 0.78 mg/L NH4 + , y 0, 0.75, 0.75 mg/L NH4 + ). Condición 5: n=1: Curva de calibrado (0, 0.28, 0.56 mg/L NH4 + ). Condición 6: n=3: Patrones de 0.375 y 0.75 (2 réplicas) mg/L NH4 + ................................ 145 Figura 55. %Recuperación de MA transformada a amonio y %Recuperación de MA no transformada, calculados respecto a la señal obtenida sin Kjeldahl, a diferentes condiciones Kjeldahl (Tabla 67). Condición 1: n=4 : 2 patrones de 0.5 mg/L y 2 patrones de 0.125 mg/L de MA. Condición 2: n=4: Patrones de MA de 0.086, 0.3875, 0.5 y 0.56 mg/L. Condición 4: n=2: Patrón de 0.5 mg/L de MA (2 réplicas). Condición 5: n=5: Patrón de 0.775 mg/L de MA (5 réplicas). Condición 6: n=2: Patrón de 1.3 mg/L de MA (2 réplicas) .............................................................................................................. 145 Figura 56. Espectro de emisión de fluorescencia de los derivados OPA-NAC a 485 nm (ëexc = 415 nm) para blanco, patrón de 0.75 mg/L de NH4 + , 1.3 mg/L MA, 1.875 mg/L EA y DMA, 2.465 mg/L N-PrA e IPA, 3.05 mg/L BA y DEA, 3.625 mg/L PeA, 4.215 mg/L HA, y 5.05 mg/L β-FEA. (56-A)-medidos antes del Kjeldahl. (56-B) medidos después del Kjeldahl ........................................................................................................... 147 Figura 57. Registros de inyección en flujo obtenidos en las condiciones experimentales óptimas a diferentes niveles de concentración de amonio (expresado en mg/L de Nitrógeno) ........................................................................................................................... 151 Figura 58. Curvas de calibrado de amonio (Señal QL frente a mg/L N) obtenidas en cinco días diferentes ........................................................................................................... 152
- 20. XVIII LISTADO DE TABLAS Tabla 1. Concentraciones típicas normales (µg/L) de agua natural, agua de río y agua marina ................................................................................................................................. 9 Tabla 2. Normativa vigente en materia de aguas ............................................................... 11 Tabla 3. Concentraciones máximas legisladas para los parámetros y tipos de agua estudiados ........................................................................................................................... 14 Tabla 4. Métodos de referencia establecidos por la legislación y métodos más utilizados recientemente ...................................................................................................................... 14 Tabla 5. Hechos históricos referidos a los fenómenos luminiscentes ................................ 18 Tabla 6. Principales reacciones quimioluminiscentes en fase gaseosa .............................. 25 Tabla 7. Principales reacciones quimioluminiscentes en fase líquida ............................... 25 Tabla 8. Principales características de algunos peroxioxalatos ......................................... 32 Tabla 9. Listado de reactivos, casas comerciales y pictogramas de seguridad .................. 34 Tabla 10. Condiciones de derivatización de aminas alifáticas .......................................... 50 Tabla 11. Parámetros de reacción estudiados .................................................................... 53 Tabla 12. Comparación entre la derivatización en cartucho y en disolución .................... 54 Tabla 13. Porcentajes de Recuperación en función del Volumen de muestra procesado .. 54 Tabla 14. Características analíticas para los derivados amina-dansilo con detección UV o fluorescente ...................................................................................................................... 57 Tabla 15. Estudios de precisión. Repetibilidad (Desviación Estándar Relativa %) .......... 57 Tabla 16. Porcentajes de recuperación y desviación estándar relativa para cada analito en las diferentes muestras de agua ...................................................................................... 59 Tabla 17. Concentraciones deducidas en la muestra de agua residual (% Error relativo) . 60 Tabla 18. Condiciones optimizadas e intervalos estudiados .............................................. 62 Tabla 19. Condiciones cromatográficas experimentales para la retención, derivatización, separación y detección en línea de aminas alifáticas ................................. 63 Tabla 20. Curvas de calibrado, parámetros analíticos y tiempos de retención para la derivatización en línea de aminas alifáticas con Dns-Cl .................................................... 64 Tabla 21. Condiciones pre y postcolumna óptimas para la determinación de aminas....... 69 Tabla 22. Parámetros analíticos de los derivados amina-dansilo detectados por quimioluminiscencia ........................................................................................................... 72 Tabla 23. Limites de detección (µg/l) obtenidos con detección quimioluminiscente a diferentes volúmenes de muestra y diferentes volúmenes de elución. Comparación con los valores obtenidos con otros sistemas de detección y reactivos ..................................... 72 Tabla 24. Estudios de precisión (DER %). Repetibilidad y reproducibilidad del método utilizando diferentes volúmenes de muestra ....................................................................... 73 Tabla 25. Concentración de amina fortificada y concentración de amina fortificada encontrada (µg/L) en muestras de agua reales .................................................................... 74 Tabla 26. Resumen de algunos procedimientos descritos en la bibliografía para la determinación de amonio .................................................................................................... 78 Tabla 27. Diseño factorial de optimización de las concentración de reactivos y pH ........ 80 Tabla 28. Importancia e interacción entre los factores: pH, [OPA] y [NAC] ................... 81 Tabla 29. Curvas de calibrado de amonio y parámetros analíticos utilizando el reactivo OPA-NAC .......................................................................................................................... 81 Tabla 30. Curvas de calibrado de amonio y parámetros analíticos utilizando el Método de Nessler ........................................................................................................................... 82
- 21. XIX Tabla 31. Concentración de amonio hallada en muestras reales utilizando los diferentes métodos estudiados ............................................................................................................. 86 Tabla 32. Curvas de adición estándar para muestras reales utilizando el Método OPA- NAC .................................................................................................................................... 87 Tabla 33. Propiedades analíticas de los tres métodos estudiados ...................................... 88 Tabla 34. Comparación del procedimiento de derivatización en disolución establecido por Marcé y col. [67] y el procedimiento de derivatización en disolución optimizado ..... 92 Tabla 35. Ecuaciones de las curvas de calibrado (Atura de pico (Voltios) frente a Concentración de NH4 + (mg/L)) obtenidas en diferentes condiciones ............................... 93 Tabla 36. Estudios de precisión (%DER) y exactitud (%Er) proporcionados por el método a diferentes niveles de concentración de amonio (n, número de réplicas) ............ 94 Tabla 37. Test t de comparación de pendientes de las ecuaciones del MOSA para todas las muestras evaluadas con la pendiente de la curva de calibrado de patrones de amonio. Los valores tabulados corresponden a un nivel de confianza del 95 % excepto *(98 %) y **(99 %) ............................................................................................................................. 96 Tabla 38. Concentración de amonio encontrada en muestras reales fortificadas con amonio a diferentes niveles de concentración antes y después del tratamiento Kjeldahl .. 97 Tabla 39. Respuesta de screening y Concentración de amonio deducida para las muestras estudiadas ............................................................................................................ 98 Tabla 40. Comparación de sensibilidad de los detectores en la reacción QL Cr(III)/luminol/H2O2 .......................................................................................................... 101 Tabla 41. Optimización de las variables químicas y físicas del sistema de inyección en flujo ..................................................................................................................................... 102 Tabla 42. Curvas de calibrado de Cu(II) y parámetros analíticos en diferentes condiciones (IL: Intervalo lineal; LD: Límite de detección) .............................................. 103 Tabla 43. Estudio de la interferencia de iones metálicos ................................................... 105 Tabla 44. Resultados obtenidos en el screening y determinación de Cu(II) en muestras de agua reales ...................................................................................................................... 106 Tabla 45. Estudios recientes basados en la reacción del luminol-H2O2 para cromo ......... 111 Tabla 46. Curvas de calibrado para Cr(III) y Cr(VI) ......................................................... 112 Tabla 47. Curvas de calibrado en el intervalo lineal para las mezclas de Cr(III)+Cr(VI) . 113 Tabla 48. Parámetros analíticos para las determinaciones de Cr(III), Cr(VI) y Cr Total .. 114 Tabla 49. Curvas de calibrado simuladas y reales utilizando modelos de calibración polinómicos ........................................................................................................................ 116 Tabla 50. Pendientes y porcentajes de recuperación para la muestra de HCl ................... 116 Tabla 51. Determinación de Cromo en una muestra de agua mineral ............................... 117 Tabla 52. Estudios de la determinación quimioluminiscente en flujo de iones metálicos en muestras de agua ............................................................................................................ 120 Tabla 53. Composición del material de referencia certificado SRM © 1640: agua dulce. 121 Tabla 54. Parámetros analíticos para la determinación de Cr(III), Co(II) y Cu(II) en muestras no acidificadas. Procedimiento en flujo .............................................................. 123 Tabla 55. Parámetros analíticos de las curvas de calibrado de patrones no acidificados utilizando el procedimiento en estático .............................................................................. 124 Tabla 56. Parámetros analíticos para la determinación de Cr(III), Co(II) y Cu(II) en muestras acidificadas con HCl 5· 10 -3 M. Procedimiento en flujo ...................................... 125 Tabla 57. Parámetros analíticos para la determinación de Cr(III), Co(II) y Cu(II) en muestras acidificadas con HNO3 0.5 M. Procedimiento en flujo ....................................... 126 Tabla 58. Modelos PLS en el intervalo lineal .................................................................... 133 Tabla 59. Modelos de PLS en el intervalo no lineal .......................................................... 134
- 22. XX Tabla 60. Modelos de PLS en condiciones ácidas ............................................................. 135 Tabla 61. Rectas de regresión lineal Valor Predicho frente a Valor Real ......................... 135 Tabla 62. Predicción para el material de referencia certificado (µg/L, n=5) ..................... 136 Tabla 63. Código y concentración del conjunto de patrones de calibración (diseño 5 2 ) ... 140 Tabla 64. Parámetros analíticos y resultados estadísticos de metilamina en presencia de cantidades variables de amonio, y amonio en presencia de cantidades variables de metilamina .......................................................................................................................... 142 Tabla 65. Características de los modelos de Regresión en Componentes Principales obtenidos para amonio y metilamina utilizando Selección Top Down (TDS), Selección del mejor Subconjunto (BSS) o Eliminación de las Variables no informativas-Selección del mejor Subconjunto ........................................................................................................ 143 Tabla 66. Concentración de amina y amonio hallada en mezclas de patrones aplicando calibración bivariada o multivariada (PCR) ....................................................................... 144 Tabla 67. Diferentes condiciones Kjeldahl ensayadas ....................................................... 146 Tabla 68. Porcentajes de recuperación de amonio para las diferentes aminas y mezclas de aminas después del Kjeldahl, con el Método OPA-NAC a 485 nm o con el Método Nessler ................................................................................................................................ 146 Tabla 69. Concentración de amina y amonio hallada en muestras reales aplicando calibración bivariada o multivariada (PCR) ....................................................................... 148 Tabla 70. Curvas de calibrado de amonio (Señal QL frente a Concentración de Nitrógeno (mg/L)) y otros parámetros analíticos en diferentes condiciones experimentales estudiadas .................................................................................................. 151 Tabla 71. Porcentajes de recuperación (bamina*100/bNH4+) para aminas alifáticas primarias y secundarias individualmente y % Recuperación medio .................................. 153 Tabla 72. Porcentajes de recuperación (bpoliamina*100/bNH4+) para poliaminas individualmente y %Recuperación medio .......................................................................... 154 Tabla 73. Porcentajes de recuperación (baminoacido*100/bNH4+) para aminoácidos individualmente y %Recuperación medio .......................................................................... 154 Tabla 74. %Recuperación individual y %Recuperación medio hallado en patrones de TEA, urea, Histidina y arginina de concentracición final de Nitrógeno de 1.5 mg/L, y para mezclas de MA/PeA/DMA (Conc final Nitrógeno 1.5 mg/L) y MA/PeA/DMA/TMA (Conc final Nitrógeno 1.8 mg/L), después del tratamiento Kjeldahl .............................................................................................................................. 156 Tabla 75. Concentración de Nitrógeno hallado en muestras de agua reales fortificadas y porcentajes de recuperación a diferentes niveles de concentración .................................... 157 Tabla 76. Concentración de Nitrógeno Total (Orgánico más amoniacal) (mg/L N) encontrada en las diferentes muestras de agua estudiadas, antes y después del tratamiento Kjeldahl ........................................................................................................... 157 Tabla 77. Principales características de los métodos propuestos en esta Tesis ................. 169 Tabla 78. Ventajas de los métodos propuestos .................................................................. 171
- 23. XXI LISTADO DE SÍMBOLOS Y ABREVIATURAS %CV Coeficiente de variación (%) %Er Error relativo (%) λem Longitud de onda de emisión λexc Longitud de onda de excitación β-FEA β-Feniletilamina ABEI Aminobutiletilisoluminol BA Butilamina Cad Cadaverina CPs Componentes Principales CTAC Cloruro de Cetil Trimetil amonio DEA Dietilamina DER Desviación estándar residual DMA Dimetilamina DNB Dinitrobenzoilo DNPO Bis(2,4-dinitrofenil)oxalato Dns-Cl Cloruro de dansilo EA Etilamina ECL Electroquimioluminiscencia EDTA Ácido Etilendiamintetracético FIA Análisis por Inyección en flujo FMOC Fluorenilmetilcloroformiato GHPSAM Método de Adición Estándar al Punto H Generalizado HA Hexilamina HPLC Cromatografía líquida de alta resolución HPLC Cromatografía Líquida de Alta Resolución IL Intervalo lineal IPA Isopropilamina IV Válvula de inyección LD Límite de detección MA Metilamina MOSA Método de Adición Estándar NAC N-Acetil-L-Cisteina N-PrA N-Propilamina OPA Orto-ftaldialdehido PCA Análisis en Componentes Principales PCR Regresión en componentes principales PeA Pentilamina PLS Regresión Mínimos cuadrados parciales PO-CL Quimioluminiscencia de los perioxalatos Put Putrescina PVA Poli Vinil Alcohol QA Química Analítica QL o CL Quimioluminiscencia QLD Detector de Quimioluminiscencia SEP Error de predicción
- 24. XXII Spd Espermidina Spm Espermina SRM Material de referencia certificado TCPO 2,4,6-triclorofenil oxalato TEA Trietilamina TKN Nitrógeno Kjeldahl Total TMA Trimetilamina TYB Blanco Total de Youden UV Ultravioleta
- 25. 1 OBJETIVOS
- 26. 2
- 27. OBJETIVOS 3 OBJETIVOS El medio ambiente constituye el sustrato que permite la supervivencia del ser humano, aportándole recursos esenciales para sus actividades económicas y productivas. La Química Analítica puede contribuir junto con otras disciplinas al estudio, vigilancia y aprovechamiento de los mencionados recursos para un desarrollo medioambientalmente sostenible. Este término se define como el conjunto de vías de progreso económico, social y político que atienden a las necesidades del presente sin comprometer la capacidad de las generaciones futuras para satisfacer sus propias necesidades. Alrededor de este término han nacido numerosas declaraciones, acuerdos, normas y leyes, introduciéndose en el seno de la Comisión mundial para el medio ambiente y el desarrollo de Naciones Unidas, más conocida como Comisión Brundtland, en 1980. La necesidad de métodos analíticos sencillos, rápidos, libres de interferencias y suficientemente sensibles para poder interpretar los datos medioambientales, controlar la contaminación y establecer el papel de cada especie en los ciclos naturales, es más que evidente. Las exigencias que le plantea el medio ambiente a la Química Analítica, se resumen en los siguientes puntos: § Análisis de muy diferentes tipos de analitos, que incluyen desde especies inorgánicas sencillas hasta complejas bio-moléculas § Matrices complejas y desconocidas, pudiendo ser líquidas, sólidas y gaseosas. § Micromuestras en algunas ocasiones y niveles de analitos del orden de trazas o inferior. § Necesidad de automatización, dado el gran número de muestras a analizar. § Necesidad de análisis in situ, por lo que requiere de equipos portátiles y robustos o sensores remotos. § Análisis de muy diferentes características. § Tratamiento de datos multivariable. Esta Tesis aporta soluciones a algunos de los puntos antes mencionados. Se proponen una serie de métodos y se revisan algunos de los ya existentes, mejorando el análisis de metales, amonio y aminas en muestras de agua medioambientales, utilizando la quimioluminiscencia como método de detección. Esta técnica puede aportar sensibilidad, selectividad y bajo coste, resultando por tanto interesante su estudio. Se pone en práctica el procedimiento analítico cubriéndose todas las etapas que lo componen, abordando tanto el muestreo como el tratamiento de las muestra, la etapa de calibración y el tratamiento y evaluación de los resultados. Los analitos han sido elegidos por sus propiedades de toxicidad y por revestir interés para el estudio de la eutrofización de aguas, lo que ha dado lugar a limitaciones en sus concentraciones que se reflejan en la legislación. Actualmente, la investigación tiende hacia la propuesta de sistemas sencillos totalmente automatizados, lo que hemos conseguido en algunos de nuestros estudios.
- 28. 4 Los proyectos concedidos al grupo de investigación y las becas disfrutadas, han sido el marco de esta Tesis: § Proyecto PB97-1387 concedido por la Dirección General de Investigación Científica y Tecnológica (DGICyT): “El método de adición estándar del punto H como método de resolución de curvas. Aplicación a determinaciones quimioluminiscentes de contaminantes ambientales”.Período de vigencia: 10-1998 a 10-2001. § Proyecto PPQ2000-1461 concedido por el Ministerio de Ciencia y Tecnología: “Métodos totales de análisis de aminas en matrices de interés medioambiental”. Período de vigencia: 28-12-2000 a 27-12-2003. § Ayudas a grupos de investigación: GR00-36 concedido por la Generalitat Valenciana: “Establecimiento de métodos globales e interpretación de datos analíticos de calidad”. Año 2000. § Ayudas a grupos de investigación: GRUPOS03-178 concedido por la Generalitat Valenciana: “Establecimiento de métodos globales e interpretación de datos analíticos de calidad”. Año 2003. § Proyecto BQU2003-6138 concedido por el Ministerio de Ciencia y Tecnología: Métodos totales de análisis de amoníaco, aminas y triazinas en matrices de interés ambiental: convencional, micro y nano”. Período de vigencia: 15-11-2003 a 14-11- 2006. § Beca predoctoral FPU (2000-2004) concedida por el Ministerio de Educación y Cultura. § Beca para la realización de una estancia breve en el Departamento de Química Analítica de la Universidad de Córdoba (período del 1 de Septiembre al 30 de Noviembre de 2002), concedida por el Ministerio de Educación y Cultura. Los objetivos específicos desarrollados se ajustan al siguiente esquema: 1. Screening de aminas alifáticas y disminución de sus límites de detección en aguas: 1.1. Preconcentración y dansilación de aminas alifáticas utilizando cartuchos C18 de extracción en fase sólida. Aplicación al análisis de screening en muestras de agua medioambientales (Apéndice 1). 1.2. Derivatización en línea de aminas alifáticas con Dns-Cl acoplada a HPLC con detección fluorescente (Apéndice 2). 1.3. Determinación quimioluminiscente de aminas alifáticas en agua mediante HPLC (Apéndice 3). 2. Disminución del límite de detección de amonio en agua e incremento de la exactitud de su determinación: 2.1. Determinación de amonio en muestras de agua utilizando el reactivo OPA-NAC. Estudio comparativo con los métodos de Nessler y electrodo selectivo de amonio (Apéndice 4). 2.2. Determinación quimioluminiscente de amonio en aguas por HPLC (Apéndice 5). 3. Método de screening de muestras para Cu(II): 3.1. Método automático de inyección en flujo para el screening de muestras de agua para Cu(II), basado en la reacción quimioluminiscente de coproporfirina I-Cu (II) / TCPO / H2O2 (Apéndice 6).
- 29. OBJETIVOS 5 4. Mejora de la exactitud de la determinación de Cr(III), Cr(VI) y Co(II): 4.1. Elaboración de una guía para la eliminación de los errores sistemáticos en la especiación de Cr(III) y Cr(VI) empleando la reacción del luminol con H2O2. Aplicación a muestras de agua (Apéndice 7). 4.2. Influencia de los protocolos de acondicionamiento de muestras de agua en la detección quimioluminiscente de elementos traza (Apéndice 8). 4.3. Calibración multivariada aplicada a la determinación quimioluminiscente simultánea de cobalto y cromo (Apéndice 9). 5. Determinación de Nitrógeno Orgánico amino y amoniacal: 5.1. Método fluorimétrico de Roth para la determinación simultánea de amonio y grupos amino primario en muestras de agua y su utilidad en la determinación del Nitrógeno Kjeldahl Total. (Apéndice 10). 5.2. Método quimioluminiscente automático para la estimación del Nitrógeno Orgánico amino (-CH2-NH-) y amonio en muestras de agua mediante la reacción del Luminol con el Hipoclorito en medio básico. (Apéndice 11).
- 30. 6
- 32. 8
- 33. Capítulo 1. INTRODUCCIÓN 9 1.1. CONSIDERACIONES SOBRE EL ANÁLISIS DE AGUA Y ANALITOS ESTUDIADOS 1.1. CARACTERÍSTICAS DE LAS AGUAS La ciencia del medio ambiente comprende el estudio completo del entorno que nos rodea y por ello necesita del apoyo de todas las ciencias, siendo por tanto multidisciplinar. Un gran número de estudios, muestran la necesidad de conocer la composición de las partes del medio implicado. No es posible por ejemplo, estudiar el transporte de sustancias en el ciclo del agua, sin medir las sustancias que están siendo transportadas. La composición química de las aguas naturales depende entre otras de variables como el tipo de agua, localización geográfica o estación del año [1]. Los constituyentes mayoritarios (1-1000 mg/L) son Na+ , Ca2+ , Mg2+ , HCO3 - , SO4 2- y Cl- , y las especies minoritarias (0.1-10 mg/L) Fe2+ , Fe3+ , Sr2+ , K+ , CO3 2- , NO3 - , F- , H3BO3. La presencia natural de otras especies ocurre en un nivel de concentraciones inferior, como se expone en la Tabla 1. Tabla 1: Concentraciones típicas normales (µg/L) de agua natural, agua de río y agua marina. Elemento Natural Río Mar Elemento Natural Río Mar Li 10 12 200 As 0.5 2 3 B 10 10 5000 Se 0.2 0.2 0.1 Al 10 50 2 Br 15 20 7000 Ti 5 10 1 Rb 1 20 120 V 0.5 1 2.5 Sr 70 60 8000 Cr 1 1 0.05 Mo 0.5 1 10 Mn 10 7 0.2 Cd 0.03 0.02 0.1 Fe 500 40 2 Sb 0.2 0.3 0.2 Co 0.05 0.2 0.02 I 2 10 60 Ni 0.5 0.3 0.05 Cs 0.02 0.04 0.1 Cu 3 5 2 Hg 0.07 0.007 0.03 Zn 15 20 10 Pb 1 3 0.03 Esta composición puede ser alterada por fuentes naturales o antropogénicas. La contaminación del medio hídrico es un problema grave y con serias consecuencias que ha afectado a numerosas zonas a pesar de la auto purificación del propio ciclo. El resultado final es que la composición de las aguas está alterada por la presencia de compuestos inorgánicos y orgánicos adicionales [1-3]. El ciclo del agua se verá afectado por el aumento de la demanda biológica de oxígeno y los procesos de eutrofización, y también por la acidificación y salinización de las aguas [4].
- 34. CONSIDERACIONES SOBRE EL ANÁLISIS DE AGUA Y ANALITOS ESTUDIADOS 10 Existen características específicas para cada tipo de agua contaminada: § Las aguas residuales urbanas e industriales son disoluciones acuosas complejas que contienen una variada gama de compuestos orgánicos e inorgánicos, tanto disueltos como en suspensión y también microorganismos [1]. Pueden contener concentraciones puntuales muy elevadas de contaminantes, lo que puede originar variaciones muy significativas en los equilibrios originando especies nuevas. § Las aguas subterráneas presentan problemas de salinidad debido a procesos de infiltración del agua marina, y también de toxicidad debido al almacenamiento de sustancias tóxicas en tanques subterráneos o a actividades agrícolas. § Las aguas costeras reciben la descarga directa de los ríos, vertidos de barcos o liberación de materiales de protección de embarcaciones. Presentan problemas de eutrofización, de bioacumulación de metales tóxicos en los moluscos o contaminación microbiana. Los problemas a niveles profundos se deben a la difusión desde la superficie o desde los sedimentos por lo que se estudian tanto gradientes horizontales como verticales, ya que ambos definen su nivel de impacto y su biodisponibilidad. Dada la gran variedad de muestras de agua medioambientales y los problemas que presentan, en esta Tesis se han estudiado diferentes tipos de agua, con el fin de abarcar una parte de la amplia variabilidad que nos podemos encontrar en cuanto a la matriz. Se han estudiado aguas: embotelladas, de grifo, de riego, de fuente, de lago, residuales y marinas. La legislación vigente en materia de aguas comprende por una parte las Directivas Europeas, y por otra, las Órdenes, Leyes y Reales Decretos tanto estatales como autonómicos. En la Tabla 2, se puede ver la normativa más representativa en los diferentes ámbitos en materia de aguas. Las normativas estatales y autonómicas se ajustan a las exigencias de la normativa europea, siendo igual o más restrictivas que ésta.
- 35. Capítulo 1. INTRODUCCIÓN 11 Tabla 2. Normativa vigente en materia de aguas. Ámbito Normativa Directiva 76/464/CEE del Consejo, de 4 de mayo de 1976, relativa a la contaminación causada por determinadas sustancias peligrosas vertidas en el medio acuático de la comunidad. Directiva relativa a aguas de baño 76/160/CEE Directiva relativa a aguas destinada al consumo humano 80/778/CEE Modificada por la directiva 98/83/CEE. Legislación comunitaria Directiv a 2000/ /CE del Parlamento Europeo y del Consejo por el que se establece un marco comunitario de actuación en el ámbito de la política de aguas. (Directiva Marco de Aguas ). Real Decreto 849/1986, de 11 de abril, por el que se aprueba el Reglamento del Dominio Público Hidráulico, que desarrolla los Títulos Preliminar, I, IV, V, VI y VII de la Ley 29/1985, de 2 de agosto, de Aguas. Real Decreto 734/1988, de 1 de julio, establece normas de calidad de las aguas de baño (BOE nº 167, de 13.07.88). Ley 22/1988, de 28 de julio, de Costas. (BOE nº 181, de 29.07.88). Real Decreto 1541/1994, de 8 de julio, que modifica el Anexo I del Reglamento de la Administración pública del agua y de la planificación hidrológica aprobado por el Real Decreto 927/1988, de 29 de Julio de 1988 Real Decreto 2116/1998, de 2 de octubre, por el que se modifica el Real Decreto 509/1996, de 15 de marzo, de desarrollo del Real Decreto-Ley 11/1995, de 28 de diciembre, por el que se establecen las normas aplicables al tratamiento de las aguas residuales. Orden de 13 de agosto de 1999 por la que se dispone la publicación de las determinaciones de contenido normativo del Plan Hidrológico de Cuenca del Júcar , aprobado por el Real Decreto 1664/1998, de 24 de julio. Real Decreto 995/2000, de 2 de junio, por el que se fijan objetivos de calidad para determinadas sustancias contaminantes y se modifica el Reglamento de Dominio Público Hidráulico, aprobado por el Real Decreto 849/1986, de 11 de abril. Real Decreto Legislativo 1/2001, de 20 de julio, por el que se aprueba el texto refundido de la Ley de Aguas (Deroga la Ley 29/1985, de 2 de agosto, de Aguas) Legislación estatal Real Decreto 140/2003, de 7 de febrero, por el que se establecen los criterios sanitarios de lacalidad del agua de consumo humano. Ley 7/1986, de 22 de diciembre, sobre utilización deaguas para riego. Ley 2/1992, de 26 de marzo, de saneamiento de las aguas residuales de la Comunidad Valenciana.Legislación autonómica Orden de 3 de junio de 2003, de la Conselleria de Agricultura, Pesca y Alimentación, por la que se establece el Programa de Actuación sobre las Zonas Vulnerables designadas en la Comunidad Valenciana.
- 36. CONSIDERACIONES SOBRE EL ANÁLISIS DE AGUA Y ANALITOS ESTUDIADOS 12 1.2. CARACTERÍSTICAS DE LOS ANALITOS ESTUDIADOS Y LEGISLACIÓN APLICABLE A LOS MISMOS. Se han estudiado analitos de diferentes tipos; metales como Cr(III), Cr(VI), Co(II) y Cu(II), amonio y nitrógeno total, aminas alifáticas primarias y secundarias (metilamina (MA), etilamina (EA), butilamina (BA), pentilamina (PeA), hexilamina (HA), dimetilamina (DMA), dietilamina (DEA) y β-feniletilamina (β-FEA)), y poliaminas (Putrescina (Put), Cadaverina (Cad), Espermina (Spm) y Espermidina (Spd)). 1.1.2.1. Descripción de los analitos estudiados Los metales se introducen en el ciclo del agua básicamente por residuos industriales. Muchos de estos iones son tóxicos para el hombre y sus emisiones han de monitorizarse y controlarse. El Cromo es un contaminante común en aguas naturales y residuales y lo podemos encontrar como Cr(III) o Cr(VI). Según el estado de oxidación de un elemento, su biodisponibilidad y toxicidad varían drásticamente, de hecho, el Cr(VI) es mucho más tóxico que el Cr(III). El Cr(III) no es tóxico a bajos niveles y se considera esencial en mamíferos. La toxicidad del Cr(VI) como aerosol es bien conocida: produce daños en la piel y sistema respiratorio, y puede producir cáncer [5]. Sin embargo, los efectos tóxicos del Cr(VI) en agua no están bien documentados. La contaminación con Cr(III) y Cr(VI) resulta de los vertidos de industrias de curtidos, acero, tintes o de sectores de fabricación de productos como pinturas, pigmentos o fungicidas. Este metal puede introducirse en aguas potables debido a los inhibidores de corrosión utilizados en cañerías o recipientes. El Cobalto se encuentra en las rocas, el suelo, el agua, plantas y animales. Existen formas radioactivas y no radioactivas de cobalto. El cobalto no radioactivo, llamado cobalto estable, se usa para producir aleaciones (mezclas de metales) usadas en la manufactura de motores de aviones, imanes, herramientas para triturar y cortar, articulaciones artificiales y también para colorear vidrio, cerámicas y pinturas y como secador de esmaltes y pinturas para porcelana. La población general está expuesta a bajos niveles de Cobalto en el aire, el agua y los alimentos. El cobalto tiene efectos tanto beneficiosos como perjudiciales sobre la salud. En bajos niveles, es parte de la vitamina B12, sustancia que es esencial para mantener una buena salud. A niveles altos, puede dañar los pulmones y el corazón [6]. El Cobre se emplea en alambres y cables eléctricos y en algunas cañerías de agua. También forma parte de aleaciones como latón y bronce. Las sales de Cobre se utilizan comúnmente en la agricultura para tratar enfermedades de las plantas, como el moho, para tratar agua, y como preservativos para alimentos, cueros y telas. Bajos niveles de cobre son esenciales para el hombre, mientras que niveles altos pueden producir efectos nocivos tales como irritación de la nariz, la boca y los ojos, vómitos, diarrea, calambres estomacales y náuseas [7].
- 37. Capítulo 1. INTRODUCCIÓN 13 El amonio es uno de los productos químicos más ampliamente utilizados. Se utiliza principalmente como fertilizante, y también en la fabricación de fibras, explosivos o plásticos y como alimento de animales. También se usa en insecticidas, en la fabricación de detergentes y limpiadores y como refrigerante [8]. Es el principal contaminante que provoca procesos de eutrofización en los sistemas acuosos, y por eso, su concentración en aguas de consumo humano viene regulada por la legislación. Las aminas alifáticas de bajo peso molecular, son intermediarios importantes en las industrias químicas y farmacéuticas. Algunas de ellas se producen en cantidades de más de 100000 toneladas por año en el Oeste de Europa. Debido a su carácter polar, son difíciles de eliminar de los sistemas acuosos. Además de su aplicación industrial, las podemos encontrar como productos de degradación de materia orgánica como las proteínas y aminoácidos u otros compuestos de nitrógeno. Las aminas alifáticas secundarias pueden reaccionar con nitrito produciendo nitrosaminas carcinogénicas. Hasta ahora, existe muy poca información acerca de la presencia de aminas alifáticas en aguas residuales industriales y en aguas superficiales, pero se sabe que en algunos tipos de agua aparecen en cantidades muy pequeñas, por lo que en este caso, los métodos para su análisis deben ser suficientemente sensibles. Las aminas biogénicas son bases orgánicas que se forman y degradan como resultado de la actividad metabólica en plantas, animales y microorganismos. Puede hallarse en fluidos biológicos, en muestras medioambientales y en corrientes de procesos industriales normalmente a niveles de trazas [9]. 1.1.2.2. Legislación aplicable Los avances en la determinación de estos u otros analitos, vendrán marcados por la legislación vigente, que con la evolución continua de los métodos de análisis, tiende a la reducción progresiva de los valores permitidos y a la exigencia de calidad en los resultados generados, priorizando la acreditación y validación de los laboratorios. La concentración de los analitos estudiados en esta Tesis, viene legislada en función del tipo de agua analizada. Para las aminas, no existe una concentración máxima permitida establecida por la legislación, pero sí lo está la concentración del Nitrógeno Kjeldahl Total (TKN). En la Tabla 3, se señalan las concentraciones máximas permitidas en cada caso. Actualmente, no existe normativa que regule la calidad exigida a las aguas superficiales destinadas a riego, por lo que se considerará la misma legislación que para aguas superficiales destinadas a potabilización. La legislación establece además los métodos de referencia para cada uno de los parámetros cuya concentración limita. En la Tabla 4, se reflejan los métodos de análisis establecidos para los analitos en estudio así como los métodos utilizados recientemente en Química Analítica en muestras de agua.
- 38. CONSIDERACIONES SOBRE EL ANÁLISIS DE AGUA Y ANALITOS ESTUDIADOS 14 Tabla 3. Concentraciones máximas legisladas para los parámetros y tipos de agua estudiados. Parámetro Tipo de agua Legislación Concentración Cromo Aguas de consumo humano Real Decreto 140/2003 50 µg/l Cobre Aguas de consumo humano Real Decreto 140/2003 2 mg/l Cobre Superficiales destinadas a potabilización Real Decreto 1541/1994 50 µg/l Cobalto - Parámetro no legislado - NH4 + Aguas de consumo humano Real Decreto 140/2003 0.5 mg/l NH4 + Superficiales destinadas a potabilización Real Decreto 1541/1994 1.5 mg/l N Kjeldahl Superficiales destinadas a potabilización Real Decreto 1541/1994 2 mg/l N Total Lagos o embalses Real Decreto 849/1986 10 mg/l N Total Vertidos de aguas residuales Orden 13 Agosto 1999 Plan Hidrográfico Júcar 20 mg/l NH4 + Aguas de baño (incluye marinas) Real Decreto 734/1988 * N Total Agua de baño (incluye aguas marinas) Real Decreto 734/1988 * *No existe un valor límite para este tipo de aguas, pero este parámetro deberá ser verificado cuando exista una tendencia a la eutrofización de aguas. Tabla 4. Métodos de referencia establecidos por la legislación y métodos más utilizados recientemente. Analito Método de referencia (Legislación) Métodos utilizados recientemente en QA Cromo Espectrofotometría de emisión por Plasma-ICP o absorción atómica (RD 995/2000) Quimioluminiscencia, absorción atómica, espectrofotometría de llama, ICP, Fluorescencia de Rayos X, MS y técnicas acopladas. Cobalto Espectrofotometría de emisión por Plasma-ICP o absorción atómica (RD 995/2000) Quimioluminiscencia, absorción atómica, espectrofotometría de llama, ICP, Fluorescencia de Rayos X, MS y técnicas acopladas. Cobre Espectrofotometría de emisión por Plasma-ICP o absorción atómica (RD 995/2000) Quimioluminiscencia, absorción atómica, espectrofotometría de llama, ICP, Fluorescencia de Rayos X, MS y técnicas acopladas. Amonio Espectrofotometría de absorción, reactivo de Nessler, método del indofenol Electrodo selectivo, métodos FIA con detección fluorimétrica o amperométrica (biosensores), quimioluminiscencia, cromatografía iónica. N Kjeldahl Método de Kjeldahl Método de Kjeldahl, Oxidación con persulfato, foto-oxidación UV, oxidación catalítica a elevadas Temp. HTCO, detección QL de Nitrógeno CLND. Aminas - HPLC
- 39. Capítulo 1. INTRODUCCIÓN 15 1.2. CARACTERÍSTICAS Y TENDENCIAS DEL ANÁLISIS MEDIOAMBIENTAL Los avances tecnológicos y metodológicos han conducido a una evolución de la Química Analítica en general y del campo del análisis del agua en particular. Este hecho se refleja en el porcentaje de publicaciones científicas (aproximadamente un 54%) que se ha dedicado a las determinaciones en matrices acuosas dentro del campo medioambiental (revisión bibliográfica en Analytical Abstract en el período 1980-2001 [10]). Los propósitos generales del análisis medioambiental [4]son la monitorización del fondo (definido como el intervalo de concentraciones en el agua de un cierto elemento, especie o sustancia derivado de fuentes biológicas, geológicas o atmosféricas naturales) y la monitorización de la contaminación (determinar la concentración de constituyentes dañinos de origen antropogénico). Los problemas que surgen en el análisis de aguas son: las bajas concentraciones de los analitos, la complejidad de la matriz, la presencia de interferentes, la variabilidad de las muestras o la especiación entre otros. En la actualidad, nos encontramos en un período de evolución de la QA medioambiental, caracterizado por el desarrollo de nuevas y mejores técnicas analíticas. El protocolo del proceso analítico dependerá del problema a analizar y requerirá un estudio de la sensibilidad, exactitud, precisión, fiabilidad, interferencias, efectos matriz, costes y tiempo de análisis, etc. teniendo siempre en cuenta factores como la validación, representatividad, impacto medioambiental y toxicidad de los reactivos. El estudio de analitos a niveles de trazas, requiere un mayor esfuerzo para garantizar la calidad del análisis. Cuanto mayores sean la precisión y exactitud exigidas, más estricto deberá ser el control de calidad. En este sentido, se han elegido los métodos quimioluminiscentes para la realización de esta Tesis puesto que proporcionan una elevada sensibilidad en un amplio intervalo dinámico de concentraciones, con una instrumentación simple (no requieren fuente de excitación), amplia versatilidad y se acoplan fácilmente como métodos de detección a sistemas de análisis por inyección en flujo o sistemas de separación cromatográficos. Como perspectiva de futuro, se han establecido una serie de analitos prioritarios para los que se deben desarrollar estudios ambientales, proponer métodos alternativos o mejorar los existentes [11]. Entre ellos, se encuentran los metales pesados, el amonio y el nitrógeno total, parámetros que han sido objeto de estudio en esta Tesis. Los avances metodológicos y químicos, vendrán marcados por la búsqueda de límites de detección cada vez más bajos y el empleo de sensores o el desarrollo de técnicas sencillas, de rápida respuesta, bajo coste y sin necesidad de personal especializado para la vigilancia. La tendencia más marcada es a la completa automatización, destacando la utilización de sistemas de inyección en flujo, así como la automatización de sistemas en HPLC. La derivatización de 7 aminas alifáticas con Cloruro de Dansilo en cartuchos C18 y posterior separación cromatográfica con
- 40. CARACTERÍSTICAS Y TENDENCIAS DEL ANÁLISIS MEDIOAMBIENTAL 16 detección fluorescente (Apéndice 1), o quimioluminiscente por reacción postcolumna de los derivados dansilados con TCPO/H2O2 (Apéndice 3), se han desarrollado en esta Tesis. Para la detección fluorescente, se ha logrado la automatización completa del análisis, consiguiendo la derivatización en línea acoplada a la separación cromatográfica (Apéndice 2). Se ha propuesto un nuevo método fluorescente para la determinación de amonio en muestras de agua, basado en la reacción del amonio con el reactivo OPA-NAC, comparándose los resultados con los obtenidos con el método de referencia de Nessler y el método del electrodo selectivo que han sido revisados previamente a su utilización (Apéndice 4). Como complemento, y con la intención de mejorar la sensibilidad, se propone un método quimioluminiscente para la determinación de amonio derivatizando este en disolución con Cloruro de Dansilo y separándolo de posibles interferentes mediante HPLC y posterior generación de quimioluminiscencia con TCPO/H2O2 (Apéndice 5). Un estudio similar se ha realizado para la determinación del Nitrógeno de grupos amino primario más Nitrógeno amoniacal, mediante la aplicación del método de fluorescencia por derivatización con el reactivo OPA-NAC (Apéndice 10), o por detección quimioluminiscente basada en la reacción del Nitrógeno Orgánico tipo amino y amonio con Hipoclorito, y medida de la disminución de señal quimioluminiscente al reaccionar con el luminol (Apéndice 11). Además, se ha extendido el uso de métodos de screening [12-13], que permiten seleccionar las muestras que superan el valor legislado, para posteriormente analizarlas por el método propuesto si está validado, o por el método de referencia que esté establecido. En esta dirección, se ha propuesto un método de análisis por inyección en flujo para el screening de muestras de agua para Cu(II), basado en la disminución de la señal quimioluminiscente emitida por el sistema Coproporfirina I/TCPO/H2O2 en presencia de Cu(II) (Apéndice 6). La especiación de elementos metálicos presenta gran relevancia debido a que las diferentes especies presentan también diferentes características en cuanto a toxicidad, biodisponibilidad o reactividad. En esta Tesis, se estudia la exactitud del método de análisis por inyección en flujo para la especiación de Cr(III) y Cr(VI) basado en la reacción quimioluminiscente del luminol con H2O2 (Apéndice 7). Como complemento a este trabajo se ha abordado también la influencia del protocolo de acondicionamiento de las muestras de agua en la detección quimioluminiscente de elementos traza (Apéndice 8). Además, basándonos en esta misma reacción, se propone un método de calibración multivariante para la determinación simultánea de Cr(III) y Co(II) en estático (Apéndice 9). Los parámetros básicos que definen un resultado analítico son la exactitud o proximidad al valor verdadero, y la incertidumbre expresada como el intervalo de confianza. Para asegurar la calidad de los resultados medioambientales existen diferentes metodologías: § Empleo de buenas prácticas de laboratorio: Consiste en realizar las distintas operaciones del laboratorio en las mejores condiciones de calidad y seguridad.
- 41. Capítulo 1. INTRODUCCIÓN 17 § Estudio de fuentes de error: Minimizar los posibles errores introducidos en las etapas del proceso analítico, evaluando la influencia en el resultado final. En el presente trabajo, y de forma general, se han evaluado las fuentes de error sistemático constante y proporcional corregibles, y se han realizado estudios de interferencias. § Control estadístico para localizar resultados anómalos. § Comparación de métodos: Estudios de confirmación: Este criterio ha sido utilizado también en esta Tesis. § Ensayos de recuperación: Se utiliza para validar resultados en muestras cuando no se dispone de un patrón certificado. Esta opción se ha seguido en la tesis de forma sistemática. § Uso de materiales de referencia certificado: Este material consiste en una muestra o patrón cuyo contenido en el analito estudiado es conocido y está avalado por un organismo competente. En los trabajos de determinación de especies metálicas, se ha utilizado un material de referencia certificado SRM ® 1640. § Ensayos interlaboratorio: Se utilizan para detectar fuentes de error, validar nuevos métodos, certificar contenidos en materiales de referencia, etc.. § Acreditación del laboratorio: Un organismo competente ha de aprobar la correcta realización de los análisis, sometiendo al laboratorio a auditorías internas y externas. Con todo esto, queda evidenciada la necesidad de nuevos métodos analíticos para poder interpretar los datos medioambientales, controlar la contaminación y establecer el papel de cada especie en los ciclos naturales.
- 42. QUIMIOLUMINISCENCIA 18 1.3. QUIMIOLUMINISCENCIA 1.3.1. PERSPECTIVA HISTÓRICA La observación e investigación de los fenómenos luminiscentes tiene una larga historia. Desde tiempos inmemoriales, se conocían sustancias y animales que resplandecían en las sombras, por lo que despertaban la curiosidad y las supersticiones [14]. En la Tabla 5, se describen los principales hechos históricos que han propiciado la investigación de este tipo de fenómenos. Tabla 5. Hechos históricos referidos a los fenómenos luminiscentes. Año Investigador Hecho 1500 -1000 a.C. Crónicas chinas Shih Ching Primeras referencias escritas sobre luciérnagas y gusanos luminiscentes (Libro de las Odas). 384 - 322 a.C. Aristóteles Observó que la luz emitida se producía sin calentamiento. 23 – 79 d.C. Caius Plinius Secundus Describió con detalle un gran número de organismos luminosos. 1555 Conrad Gesner Publicó un libro cuyo contenido trataba exclusivamente de los fenómenos luminiscentes. 1565 Nicolás Monarde Escribió acerca del extraordinario color azul intenso de un extracto acuoso de la madera llamada “lignum nephrilicum”. 1603 Vincenzo Cascariolo Introdujo la luminiscencia de los sólidos. Calentó polvos de sulfato de bario con carbón y encontró que la mezcla resultante brillaba en la noche. La piedra se “cargaba” de luz solar por el día y brillaba durante horas en la oscuridad (fosforescencia). Le llamó lapis solaris (piedra del Sol). 1652 Nicolas Zucchi Demostró, por medio de filtros ópticos, que el color de la luz emitida durante la noche era la misma que cuando la piedra era expuesta a luz blanca o de otros colores, como azul o verde. 1655 Athanasius Kircher, Francisco Grimaldi, Isaac Newton y Robert Boyle Observaron que cuando el extracto acuoso de la madera “lignum nephrilicum” era ilu minada con luz blanca, aparecía una luz reflejada azul intensa, mientras que la luz transmitida era amarilla. 1668 Robert Boyle Marcó inicialmente las diferencias esenciales entre incandescencia y luminiscencia (emisión de luz por materiales calentados o no calentados respectivamente). 1845 George Stokes Caracterizó la naturaleza bicromática del cristal de fluoruro cálcico como una emisión (fosforescencia). Demostró que la luz incidente de una región espectral era absorbida y transformada por la solución en luz emitida en una región espectral diferente, de mayor ë (Ley de Stokes). La emisión desaparecía instantáneamente cuando se apagaba la luz incidente. Le dio el nombre de fluorescencia.
- 43. Capítulo 1. INTRODUCCIÓN 19 1877 Radziszewski Observó por primera vez la emisión de luz causada por reacciones químicas [15] (QUIMIOLUMINISCENCIA) al burbujear con O2 una disolución etanólica alcalina de lofina. 1888 Eilhard Wiedemann Introdujo el término luminiscencia para describir la emisión de luz que no requería un aumento de la temperatura, abarcando la fluorescencia y la fosforescencia. 1927 Albrecht Describió las propiedades quimioluminiscentes del luminol[16]. 1935 Gleu y Petsch Describieron la luz azul o verde emitida por el nitrato bis(N-Metilacridinio) (lucigenina) [17]. 1964 McCapra Propuso un mecanismo basado en la formación de un ciclo de dioxoetanona para explicar la quimioluminiscencia de las sales acridínicas [18]. Hoy en día, la luminiscencia se entiende como el proceso por el cual un material genera radiación no térmica (depende de las características del tipo de material) [19]. Así, la luminiscencia es la emisión de luz por medios diferentes a la combustión y por eso ocurre a temperaturas más bajas que las requeridas por la combustión. La intensidad de emisión luminiscente es baja si se compara con las fuentes incandescentes, pero tiene la ventaja de que habitualmente emana de cantidades de materia extremadamente pequeñas. Esto proporciona implicaciones beneficiosas para las ciencias analíticas, sin embargo, el uso de la luminiscencia para el análisis es bastante reciente. Registrando la intensidad de luminiscencia relativa en función de la concentración, se pueden determinar cuantitativamente (frecuentemente a niveles de trazas) un amplio rango de analitos tanto inorgánicos como orgánicos. La luminiscencia, generalmente ofrece superior selectividad, detectabilidad y rango lineal comparado con la espectrometría de absorción. Muchas publicaciones analíticas muestran que la metodología fluorimétrica es más común que la fosforescencia o la quimioluminiscencia. Sin embargo, los trabajos de investigación indican un aumento en la aceptación de estas dos últimas técnicas espectroscópicas, específicamente cuando se combinan con separaciones cromatográficas [20-24]. Las investigaciones sobre el potencial analítico de la quimioluminiscencia (QL) para análisis de rutina datan de los años 70 para las reacciones en fase gaseosa, y de la década de los 80 para las reacciones en fase líquida. Hoy en día el interés analítico en la aplicación de la QL está creciendo exponencialmente.
- 44. QUIMIOLUMINISCENCIA 20 1.3.2. FUNDAMENTOS Y MECANISMOS La QL se define como la emisión de radiación electromagnética, normalmente en la región del visible o infrarrojo cercano, producida por una reacción química. Para que se dé la QL es necesario que la reacción produzca un exceso de energía, lo cual es bastante frecuente sobretodo en reacciones rédox, pero el hecho de que este exceso de energía se disipe con emisión quimioluminiscente, depende en gran medida de la estructura molecular de los intermedios o productos de reacción. Las reacciones quimioluminiscentes pueden generarse mediante dos mecanismos básicos, por lo que la clasificación de estas reacciones se ha establecido en función de estos dos mecanismos: - Quimioluminiscencia directa: Se parte de dos reactivos, A y B, normalmente un substrato y un oxidante. A y B reaccionan dando un intermedio de reacción en un estado electrónicamente excitado. Este intermedio, al relajarse hasta el estado fundamental, emite un fotón. A veces, en estas reacciones, se requiere un catalizador que disminuya la energía de activación y aumenta el rendimiento cuántico de la reacción quimioluminiscente. - Quimioluminiscencia indirecta (sensibilizada o de transferencia de energía): El mecanismo es el mismo para la formación del intermedio electrónicamente excitado, pero este intermedio no puede emitir directamente el fotón para dar la QL y requiere la presencia de un fluoróforo al cual le transfiere la energía, de forma que el fluoróforo se excita y al volver a su estado fundamental emite un fotón. En la Figura 1, se resumen esquemáticamente los dos mecanismos descritos: Figura 1. Mecanismos de las reacciones quimioluminiscentes. A + B (Precursor QL) (Oxidante) Catalizador P* (Intermedio o producto electrónicamente excitado) QL QL DIRECTA F NDIRECTA P + hνν P + F* F + hνν
- 45. Capítulo 1. INTRODUCCIÓN 21 Por tanto, los procesos quimioluminiscentes comprenden los siguientes pasos: 1. Reacción química inicial que proporciona el intermedio o producto. 2. Conversión del exceso de energía química en excitación electrónica de este intermedio. 3. Transferencia de energía en el caso de la QL indirecta. 4. Emisión de la luz por parte de las especies excitadas. El rendimiento cuántico de una reacción quimioluminiscente, dependerá por tanto de los rendimientos cuánticos de los cuatro pasos implicados en los mecanismos. 1.3.3. CARACTERÍSTICAS DE LA QUIMIOLUMINISCENCIA COMO TÉCNICA ANALÍTICA 1.3.3.1. Ventajas y limitaciones de la técnica En las reacciones quimioluminiscentes, la intensidad de emisión es función de la concentración de las especies químicas implicadas, por lo que las medidas de intensidad de emisión pueden emplearse con fines analíticos. Las principales ventajas de los sistemas quimioluminiscentes que los hacen adecuados para el análisis cuantitativo en Química Analítica son: - Elevada sensibilidad y amplio intervalo dinámico de concentraciones, llegando en algunos casos a límites de detección del orden de los femtomoles. - No requiere fuente de excitación externa, lo que se traduce en ausencia de dispersión y de señales de fondo, en la reducción de interferencias debidas a procesos de excitación no selectivos y en la utilización de una instrumentación más sencilla que otros procesos luminiscentes. - Versatilidad: Se puede utilizar la técnica para determinar cualquiera de las especies que participan en el proceso quimioluminiscente: sustratos, catalizadores, inhibidores, fluoróforos, especies que dan reacciones acopladas aumentando o disminuyendo la concentración de los reactivos implicados en la reacción quimioluminiscente. - Se acoplan fácilmente como método de detección en cromatografía, electroforesis capilar o inmunoensayo. Entre las limitaciones de estas reacciones para el análisis, los siguientes parámetros se han de tener perfectamente controlados: - Los factores experimentales que afectan al rendimiento cuántico y la velocidad de reacción son: la estructura química del precursor quimioluminiscente, la naturaleza y concentración de sustancias que favorecen procesos competitivos no radiantes, el
- 46. QUIMIOLUMINISCENCIA 22 catalizador elegido, la temperatura, la fuerza iónica, la presencia de aceptores de la energía transferida, la presencia de iones metálicos, la hidrofobicidad del disolvente y la composición de la disolución. - El tiempo de medida de la señal, debido a que la emisión quimioluminiscente varía en función del tiempo. Tras la mezcla de los reactivos, la emisión de luz alcanza inmediatamente un máximo y después cae hasta la línea base en un rápido destello menor que un segundo o en emisión continua que puede durar desde minutos a horas. En la Figura 2, se muestra un ejemplo de la variación de la señal quimioluminiscente con el tiempo, para la reacción del luminol/H2O2 estudiada en esta Tesis, utilizando Cr(III) como catalizador (Concentración final Cr(III) 5.5 µg/l). Como se puede observar, la emisión es muy rápida, aproximadamente 1s, y por tanto, el tiempo al que se registre la señal será muy decisivo. Figura 2. Intensidad de emisión quimioluminiscente frente al Tiempo. 0 1 2 3 4 5 Tiempo (segundos) 0 100 200 300 Intensidaddeemisión
- 47. Capítulo 1. INTRODUCCIÓN 23 1.3.3.2. Tendencia Actual La importancia de las determinaciones quimioluminiscentes en el campo de la Química Analítica, ha tenido una evolución creciente en los últimos años. Durante los años 70 y 80, la investigación en este tipo de reacciones estaba orientada a la búsqueda de nuevos reactivos quimioluminiscentes [25-27] y el estudio de reactivos estructuralmente semejantes a los que ya se conocían como quimioluminiscentes. A partir de los años 90 y hasta la actualidad, los estudios de reacciones quimioluminiscentes se han dirigido más hacia la mejora en la instrumentación, construyendo detectores muy sencillos para este tipo de medidas [28-30], y también a la obtención de estos sistemas de detección acoplados a técnicas separativas permitiendo la resolución y cuantificación de varios analitos en mezclas complejas por ejemplo de ácidos[31], compuestos de nitrógeno [32], o biopolímeros [33]. Las grandes aplicaciones de la QL como método de detección en flujo, cromatografía líquida o electroforesis capilar, junto con el gran potencial del inmunoensayo, hacen de la técnica un campo de investigación muy interesante en una amplia variedad de disciplinas. La detección quimioluminiscente es una técnica en pleno desarrollo. El interés de la QL en Química Analítica, queda reflejado en el número de publicaciones científicas en revistas de gran prestigio (Analytical Chemistry, Analytica Chimica Acta, The Analyst, Talanta, Journal of Chromatography y Journal of pharmaceutical and Biomedical Analysis entre otras) que, según la base de datos Analytical Abstracts desde 1979 hasta la actualidad (finales de 2003), es del orden de 3700 publicaciones. La Figura 3 refleja el crecimiento exponencial en el número de publicaciones durante estos últimos 25 años. Figura 3: Número de publicaciones en revistas científicas de ámbito internacional frente a cada año desde 1979 a 2003. 0 100 200 300 1979 1982 1985 1988 1991 1994 1997 2000 2003 Año NºPublicaciones
- 48. QUIMIOLUMINISCENCIA 24 1.3.4. REACCIONES QUIMIOLUMINISCENTES La bibliografía muestra la aplicación de reacciones quimioluminiscentes en fase sólida, líquida y gaseosa. Las reacciones en fase sólida han sido las menos aplicadas pues son reacciones muy limitadas por la débil emisión que acompaña a la oxidación de muchos materiales orgánicos. Los sólidos se calientan en presencia de oxígeno y la emisión QL es proporcional a la velocidad de reacción. En ausencia de oxígeno, la emisión QL se debe a la descomposición de los peróxidos formados. Se puede recoger la emisión directa de estas especies o la indirecta (exaltando la QL por adición de fluoróforos). En este campo, se han descrito reactores para la detección quimioluminiscente en fase sólida, utilizando como soportes metacrilatos polimerizados. Un ejemplo es la reacción de perioxalato con peróxido de hidrógeno que utiliza como fluoróforo el 3-aminofluoranteno inmovilizado en el soporte [34]. Los métodos quimioluminiscentes en fase gaseosa, se han utilizado combinados con cromatografía gaseosa (utilizando por ejemplo la reacción quimioluminiscente del ozono [35]), cromatografía de fluidos supercríticos (reacciones del nitrógeno y el sulfuro [36-38]), y también con detectores de gases no cromatográficos en los que se requiere una respuesta rápida como el detector quimioluminiscente de monitorización continua de sulfuro [39]. Algunos de los analitos que se han determinado mediante esta técnica son: óxidos de nitrógeno (NOx), ozono, y peróxido de hidrógeno en muestras atmosféricas, y detección y secuenciación de DNA, ácidos nucleicos, fosfatasa alcalina yoxidantes en riñones, cerebro y plasma. En la Tabla 6, se pueden observar las principales reacciones en fase gas [40] y las condiciones de reacción necesarias en cada caso, siendo las más aplicadas las utilizadas para la determinación de azufre, fósforo y nitrógeno total. En algunos casos la reacción se da a temperatura ambiente, y en otros, se requiere calefacción a elevadas temperaturas. Las reacciones quimioluminiscentes en fase líquida, tienen aplicabilidad en la determinación de un amplio rango de analitos, desde elementos metálicos como cromo y cobalto a niveles traza [41-42], hasta fármacos como tiopronina [43] o azitromicina [44]. En la Tabla 7, se reflejan las principales reacciones quimioluminiscentes utilizadas en fase líquida para diferentes analitos estudiados [40,45]. Se dividirán estas reacciones según la QL sea directa o indirecta. Dado que las reacciones quimioluminiscentes estudiadas en esta Tesis, son en fase líquida, se detallará más ampliamente este apartado.
- 49. Capítulo 1. INTRODUCCIÓN 25 Tabla 6. Principales reacciones quimioluminiscentes en fase gaseosa. Analito Reactivo λλem (nm) Reacción Temperatura Azufre H2 384-394 S + S → S2 * S2 * → S2 + hν Llama H2 Compuestos Fósforo H2 526 Comp-P → HPO* HPO* → HPO + hν Llama H2 O3 Alquenos 300-600 Alqueno + O3 → CO CO + O3 → CO2 * + O2 → CO2 + hν 25ºC Hidro - Carburos O3 300-600 HC + O3 → CO CO + O3 → CO2 * + O2 → CO2 + hν 150-200ºC NO O3 590 NO + O3 → NO2 * + O2 NO2 * → NO2 + hν 25ºC Compuestos Nitrógeno O2/O3 590 NO + O3 → NO2 * + O2 NO2 * → NO2 + hν 800-1000ºC Compuestos Sulfurados H2 260-480 Comp-S → SO SO + O3 → SO2 * + O2 SO2 * → SO2 + hν Llama H2 Tabla 7. Principales reacciones quimioluminiscentes en fase líquida. Reacción Analitos λλmáx, em Rendim. cuántico* Oxidación del luminol en medio básico acuoso Catalizadores (Cr(III), Co(II),...), H2O2, luminol.. 425 nm 0.01 Oxidación del luminol en dimetilsulfóxido Superóxido 500 nm 0.05 Oxidación de la lucigenina con peróxido de hidrógeno alcalino Cu(II),Pb(II), Hg(II), I- , ... 440 nm 0.016 Oxidación de lofina en medio etanólico con hidróxido sódico Na+ , Mg2+ , lofina 525 nm - Oxidación de perioxalatos Proteínas, Cl2 , porfirinas, derivados del DnsCl,... Depende del sensibilizador 0.05-0.5 Reducción de tris(2,2’-bipiridil)rutenio(III) Aminas, Aminoácidos, Alcaloides, ión oxalato,... 610 nm - Oxidación de alcaloides con permanganato potásico en medio ácido en presencia de polifosfatos Alcaloides, anestésicos, aminoácidos, indoles, ácido ascórbico, fenoles, .. 680 nm - Oxidación de D-luciferina con luciferasa dependiente del ATP pH 8.6 ATP, enzima luciferasa, Mg(II),... 560 nm 0.88 *La intensidad de emisión de una reacción depende del rendimiento cuántico, que es una medida de la eficiencia de la reacción quimioluminiscente. Los rendimientos cuánticos van desde 1· 10-15 (QL ultradébil) hasta cerca de 1 (procesos bioluminiscentes).
- 50. QUIMIOLUMINISCENCIA 26 1.3.5. REACCIONES QUIMIOLUMINISCENTES DIRECTAS EN FASE LÍQUIDA 1.3.5.1. Reacciones del Luminol y sus análogos El luminol (5-amino-2,3-dihidro-1,4-ftalazindiona), es uno de los reactivos quimioluminiscentes más comúnmente empleado en fase líquida. Fue presentado por primera vez en 1928, y la caracterización de la cinética de reacción y las especies emisoras requirió más de tres décadas de investigación. El luminol se oxida casi cuantitativamente a 3-aminoftalato en medio básico tanto en disolventes polares como apolares. El mecanismo propuesto para la reacción [45] es el que se muestra en la Figura 4. Figura 4. Mecanismo propuesto para la reacción del luminol. Como oxidantes de la reacción, se han utilizado: el permanganato en la determinación de paracetamol en fármacos [46] o de urea en tejidos de plantas [47]; el hipoclorito, en la determinación de amonio [48] o catecolaminas [49]; y el yodo, en la determinación de penicilinas [50-51]; pero el oxidante más utilizado ha sido el peróxido de hidrógeno. O2 O ONH2 NH NH OH- O2 O ONH2 N N O ONH2 N N O O - - + N2 O ONH2 O ONH2 O O - - * O ONH2 O ONH2 O O - - + hν (λ = 425 nm) Luminol 3-aminoftalato
- 51. Capítulo 1. INTRODUCCIÓN 27 Cuando la reacción del luminol con peróxido de hidrógeno se da en medio orgánico (en dimetilsulfóxido), no se requiere ningún catalizador, pero cuando se da en medio acuoso, se requiere la presencia de un catalizador. Como catalizadores, se han utilizado diferentes metales de transición (Co2+ , Cu2+ , Fe3+ , Cr3+ ,...) [52-54], ferricianuro [55-56], o algunos metalocomplejos (hemina, hemoglobina y peroxidasas [57-58]). La intensidad de emisión quimioluminiscente tiene un máximo a 425 nm cuando se trabaja en medio acuoso, y a 500 nm cuando se trabaja en dimetilsulfóxido. El pH óptimo de la reacción está entre 8 y 11, dependiendo del catalizador. La reacción se ha utilizado para la determinación de los catalizadores (límites de detección desde 1 µM hasta 1 nM según las especies), del H2O2 (límite de detección 0.1 µM) o del luminol (límite de detección 1 pM) [59-60]. También se puede iniciar la reacción QL electroquímicamente, sobre un electrodo de Ag/AgCl a +0.5V [61-62]. Los límites de detección con QL electrogenerada son similares a los obtenidos con un catalizador convencional. En los últimos años, se han revisado compuestos de estructura similar al luminol con la intención de mejorar el rendimiento cuántico, y se ha concluido que la menor alteración en el anillo heterocíclico destruye las propiedades quimioluminiscentes de la molécula. El luminol se puede enlazar a algunos analitos para inmunoensayo mediante la sustitución de los hidrógenos del grupo amino primario; esta sustitución provoca una disminución en la intensidad de emisión quimioluminiscente. Compuestos derivados del luminol como el isoluminol (4-aminoftalhidrazida) [63-64] también presentan propiedades QL pero son menos eficaces. Otro derivado del luminol, el ABEI (aminobutiletilisoluminol) [65-66] se utiliza como reactivo marcador. En esta Tesis, se presentará una guía para evitar los errores sistemáticos en la determinación quimioluminiscente de Cr(III) y Cr(VI) en medio acuoso, utilizando la reacción del luminol con H2O2 y un sistema de análisis por inyección en flujo (Apéndice 7). Se revisará también la influencia de los protocolos de acondicionamiento de la muestra en el análisis quimioluminiscente de elementos metálicos a niveles de trazas (Apéndice 8). Por otra parte, se aplicará esta misma reacción en estático, proponiendo un modelo de calibración multivariada para la determinación quimioluminiscente simultánea de Cr(III) y Co(II) (Apéndice 9). En todos los casos, se abordará el análisis de muestras de agua. También se propone la utilización de la reacción del luminol con Hipoclorito sódico como oxidante en medio básico. Se determinará la cantidad de Nitrógeno Orgánico (aminas y aminoácidos) y Amoniacal en muestras de agua basándose en la disminución de la señal QL por reacción de los compuestos de nitrógeno con hipoclorito para dar cloraminas (Apéndice 11). La determinación QL se comparará con la estimación del Nitrógeno Kjeldahl Total por digestión-destilación y detección
- 52. QUIMIOLUMINISCENCIA 28 fluorescente basada en la reacción del amonio con el reactivo OPA/NAC o UV/vis basada en la reacción del amonio con el reactivo de referencia de Nessler (Apéndice 10). 1.3.5.2. Reacción de los ésteres de acridinio Los ésteres de acridinio reaccionan con peróxido de hidrógeno en medio alcalino dando lugar a N-Metilacridona en estado excitado que emite luz a 440 nm. El mecanismo propuesto para esta reacción [40] se muestra en la Figura 5, no se requiere de catalizadores. Se ha utilizado para la determinación cuantitativa de peróxido de hidrógeno [67], pero la principal aplicación de estos ésteres radica en su uso como marcadores en inmunoensayos [68] y estudios de DNA [69-70]. Los máximos rendimientos cuánticos en la emisión QL de los ésteres de acridinio se obtienen a pH 12-13, pero a estos pHs los ésteres de acridinio se descomponen dando una pseudo-base no-quimioluminiscente. Por esto, el H2O2 se añade en medio ácido para reconvertir la pseudo-base a éster de acridinio y posteriormente se añade el hidróxido sódico para iniciar la reacción QL. Al igual que en la reacción del luminol, también se puede generar el H2O2 electroquímicamente. Figura 5. Mecanismo de la reacción QL de los ésteres de acridinio. La lucigenina, nitrato de bis-N-acridinio, no lleva el grupo éster, pero sigue el mismo mecanismo que los ésteres, con la única diferencia que requiere de la presencia de iones metálicos de transición que actúan como catalizadores. Por esta razón, se puede utilizar también la reacción para la determinación de iones metálicos si se utiliza lucigenina como reactivo quimioluminiscente. N + CH3 C O O R OH, H2O2 N CH3 O * Ester de acridinio N-Metil acridona* NMA* NMA + h ν
- 53. Capítulo 1. INTRODUCCIÓN 29 1.3.5.3. Reacción de Tris (2,2’-bipiridina) rutenio (III) Las propiedades fotoluminiscentes de los N’N’ quelatos de Ru(II) fueron presentadas por primera vez en 1959, y posteriormente en 1962, se observó la QL emitida por el Tris(2,2’-bipiridina) rutenio (III). Sin embargo, hay muy poca información acerca del mecanismo de la reacción. El estado excitado del complejo Ru (bipi)3 2+ , produce una emisión naranja a 610 nm. La emisión es fosforescencia químicamente inducida. Se puede llegar a este estado excitado de diferentes formas [45]: § Ru(bipi)3 3+ + Ru(bipi)3 + → [Ru(bipi)3 2+ ]* § Ru(bipi)3 + + Oxidante → [Ru(bipi)3 2+ ]* + Oxidante- § Ru (bipi)3 3+ + Reductor → [Ru(bipi)3 2+ ]* + Reductor+ La intensidad de quimioluminiscencia en estas reacciones es proporcional a la concentración de cualquiera de los reactivos. Como reductores se pueden utilizar una gran variedad de compuestos, pero no todos producen la típica QL naranja. Algunos que sí la producen son: aminas [71-72], aminoácidos [73], alcaloides [74-75] e ión oxalato [76-77], a concentraciones incluso por debajo de 1· 10-10 M. La reacción es tan selectiva que incluso para alcaloides como la codeína y la morfina con una estructura química tan similar, la codeína se puede determinar, pero la morfina no da QL. Esto ha dado lugar a que no existan todavía mecanismos definitivos para este tipo de reacción. Para las aminas, la respuesta aumenta con el número de sustituyentes (terciarias > secundarias > primarias). Recientemente, se ha estudiado un complejo similar Ru(Phen)3 2+ (Phen: 1,10- fenantrolina), que presenta mayor emisión al oxidarse con Ce(IV) en medio sulfúrico. 1.3.5.4. Reacciones Bioluminiscentes Los sistemas más utilizados se basan en la reacción de luciferina-luciferasa [78- 79]. La enzima luciferasa (muy específica) cataliza la oxidación por el aire de la luciferina en presencia de ATP, que se consume como substrato y emite luz a 562 nm. Para esta reacción es necesaria la presencia de Mg(II) para que la luciferasa muestre su actividad. Por cada molécula de ATP consumida, se emite un fotón, por esto, esta reacción es un sistema analítico ideal para detectar la presencia de ATP.
- 54. QUIMIOLUMINISCENCIA 30 Este sistema se emplea en el ámbito de la gestión y garantía de calidad para aumentar la higiene en alimentos, en control de calidad ambiental, en el seguimiento de plantas de aguas residuales y en la industria farmacéutica, entre otros. 1.3.5.5. Reacciones de oxidación directa Existen reacciones quimioluminiscentes por combinación de una especie dada con oxidantes y reductores fuertes en diferentes condiciones químicas. Se han utilizado estas reacciones para la determinación de aminoácidos, SO2 o morfina con MnO4 - en medio ácido y alcalino [80-81], ácido ascórbico con Ce(IV) [82], nitrito con H2O2 [83], tetraciclina con Br2 [84], entre otras aplicaciones. Otros oxidantes utilizados habitualmente son el ClO- y el IO4 - . Normalmente, cuando la oxidación o la reducción del analito da un producto fluorescente o el analito en sí presenta fluorescencia, cabe la posibilidad de que la oxidación del analito produzca emisión QL. Para aumentar la emisión QL en estos casos, se han añadido al medio de reacción sensibilizadores, medios micelares o catalizadores. Estas reacciones, se han aplicado principalmente a métodos de inyección en flujo con detección quimioluminiscente. 1.3.6. REACCIONES QUIMIOLUMINISCENTES INDIRECTAS EN FASE LÍQUIDA 1.3.6.1. Reacciones con dioxoetanos Los dioxoetanos son peróxidos cíclicos de 4 miembros. Muchos de ellos son inestables y se han descrito como intermediarios en muchas reacciones quimioluminiscentes. Su estabilidad relativa depende de los grupos sustituyentes. A temperatura ambiente, los 1,2-dioxoetanos sustituidos son estables, pero pueden descomponerse químicamente produciendo la QL. Los 1,2-dioxoetanos utilizados en las reacciones quimioluminiscentes tienen un grupo adamantil en una parte del anillo, y un grupo arílico sustituido (ArilO-X) en la otra. La estabilidad de esta molécula se pierde al añadir un reactivo que provoca una reacción de transferencia de carga eliminando el grupo X que estabilizaba la estructura, dando lugar a su descomposición espontánea que proporciona un éster arílico en estado excitado. El mecanismo detallado [40] se puede ver en la Figura 6. La emisión QL la darán, tanto la especie arílica excitada de forma directa, como un fluoróforo añadido a la mezcla de reacción al cual le transfiere la energía (QL indirecta). Esta reacción, se ha utilizado para la determinación de dioxoetanos como los 5-tert-butil-1-(4-hidroxibenz[d]oxazol-6-il)-4,4-dimetil-2,6,7-trioxabiciclo[3.2.0]heptanos [85]; para la determinación de los fluoróforos, como β-dicetonatos de praseodimio (III), neodimio (III) e Yterbio (III) excitados en la descomposición de 1,2-dioxoetano [86]; o
- 55. Capítulo 1. INTRODUCCIÓN 31 para la determinación del reactivo utilizado en la reacción de descomposición del 1,2- dioxetano como el enzima proteasa [87]. Figura 6. Mecanismo de la reacción QL de dioxoetanos. 1.3.6.2. Reacciones con peroxioxalatos En QL indirecta, el sistema más frecuentemente utilizado se basa en la reacción quimioluminiscente de los peroxioxalatos (PO-CL). En estas reacciones, un éster aril oxalato es oxidado por peróxido de hidrógeno en presencia de un fluoróforo. El mecanismo propuesto [45] utilizando bis-(2,4,6-triclorofeniloxalato) (TCPO) se muestra en la Figura 7. En la reacción, se forma un intermedio de elevada energía, la 1,2- dioxoetanodiona, que forma un complejo de transferencia de carga con el fluoróforo; el fluoróforo cede un electrón al intermedio y el complejo se disocia, alcanzando el fluoróforo un estado excitado. La emisión se produce al volver el fluoróforo al estado fundamental. Por tanto, las mejores condiciones de emisión quimioluminiscente se obtendrán cuanto más fácilmente oxidable sea el fluoróforo. Figura 7. Mecanismo propuesto para la reacción QL con peroxioxalato. Cl Cl Cl O C O C O O Cl Cl Cl + H2O2 Cl Cl Cl O C O C O O OH O C O C O O F O C O C O O .- F .+ F* + 2CO2 F + hν O- O CH3O * O O OCH3 OX O O OCH3 O- hν
- 56. QUIMIOLUMINISCENCIA 32 Puede añadirse a la reacción una base débil como trietilamina o imidazol, para catalizar la reacción y aumentar la intensidad de QL obtenida. La reacción se da en un amplio rango de pH, desde 5 a 9, pero generalmente se utiliza un pH cercano a la neutralidad. El principal inconveniente de la reacción es la limitada solubilidad y estabilidad de los peroxioxalatos en medio acuoso. Generalmente se utilizan disolventes orgánicos para evitar problemas de degradación del reactivo. En la Tabla 8, se pueden ver las características de algunos de los ésteres de oxalato [15] utilizados en esta reacción. La QL total es el área bajo la curva de emisión. De estos ésteres, los más utilizados son el TCPO y el DNPO. Tabla 8. Principales características de algunos peroxioxalatos. Reactivo quimioluminiscente QL Total Tiempo de emisión (s) Bis(2,4-dinitrofenil)oxa lato DNPO 7500 11 Bis(2,4,6-triclorofenil)oxalato TCPO 8990 23 Bis(2-(3,6,9-trioxadeciloxicarbonil)-4- Nitrofenil)oxalato TDPO 9370 13 Bis(2,6-fluorofenil)oxalato DFPO 5650 97 Bis(2-(3-oxabutiloxicarbonil)-4- Bromofenil)oxalato MBO-1 4630 630 Bis(2-(3,6-dioxaheptiloxilcarbonil)-4- Bromofenil)oxalato MBO-2 5000 750 Este tipo de reacciones, se ha utilizado para la determinación cuantitativa de H2O2 obteniendo límites de detección del orden de 0.18 pM [88]. La aplicación analítica más común, ha sido la detección en cromatografía líquida de fluoróforos o compuestos que no son fluorescentes pero pasan a serlo al derivatizarlos con cloruro de dansilo (como aminoácidos [89, 23], esteroides [90], aminas alifáticas [91] y ácidos carboxílicos [92] entre otros). Para fluoróforos fácilmente oxidables tales como los derivados del dansilo, la detección quimioluminiscente proporciona mayor sensibilidad que la fluorescente, debido a que no existe dispersión debida a la fuente de excitación. En esta Tesis, la reacción con peroxioxalatos y concretamente, la reacción del TCPO/H2O2, se ha aplicado a la determinación de Cu(II). La Coproporfirina I actúa como fluoróforo en la reacción del peroxioxalato. En presencia de Cu(II), la intensidad de emisión disminuye por formación del complejo Cu(II)-Coproporfirina I. Basado en esta reacción de inhibición, se ha diseñado un sistema FIA para el screening de Cu(II) en muestras de agua (Apéndice 6). Además, se ha utilizado esta misma reacción para la determinación cuantitativa y screening de NH4 + en muestras de agua mediante HPLC. Para ello, se ha derivatizado en disolución el amonio con cloruro de dansilo, de forma que el derivado actúa de
- 57. Capítulo 1. INTRODUCCIÓN 33 fluoróforo en la reacción quimioluminiscente. El derivado se inyecta en un sistema cromatográfico y posteriormente se mezcla con TCPO y H2O2 en un sistema post- columna (Apéndice 5). El método se compara con otros tres métodos estudiados, el método de referencia de Nessler, el método del electrodo selectivo, y un método fluorescente basado en la reacción del amonio con el reactivo OPA/NAC (Apéndice 4). Finalmente, se ha aplicado también esta reacción a la determinación cuantitativa de aminas alifáticas en aguas mediante HPLC, derivatizando las aminas con cloruro de dansilo en cartucho C18 de extracción en fase sólida, metodología introducida por primera vez por el grupo en 1996. La mezcla de derivados de aminas se inyecta en el sistema cromatográfico donde se separan los derivados y posteriormente se mezclan con los reactivos TCPO y H2O2 en un sistema post-columna (Apéndice 3). Hemos comparado los resultados obtenidos utilizando detección quimioluminiscente con los obtenidos mediante la detección directa del derivado tanto por fluorescencia como por UV/vis (Apéndice 1). Se compara también con un sistema totalmente automatizado en el que la derivatización con cloruro de dansilo se lleva a cabo en línea utilizando detección fluorescente (Apéndice 2).
- 58. REACTIVOS 34 1.4. REACTIVOS En la Tabla 9, se muestra el listado de reactivos utilizados para la realización de esta Tesis, las casas comerciales que los suministraron y los pictogramas de seguridad de cada uno de ellos. Tabla 9. Listado de reactivos, casas comerciales y pictogramas de seguridad. Reactivo Casas Comerciales Xi Xn T/T+ I/I+ C N β-Feniletilamina Sigma X 1,7-diaminoheptano Sigma X 2,4,6-triclorofenil oxalato Fluka X Ác. Etilendiamintetracético Probus Acetato amónico Panreac Acetona Scharlau +X Acetonitrilo Scharlau, J.T.Baker X +X Ácido aspártico Aldrich Ácido bórico Scharlau X Ácido clorhídrico Fluka, Merck X Ácido glutámico Aldrich Ácido nítrico Merck X Ácido sulfúrico Fluka, Merck X Alanina Aldrich Albúmina bovina Fluka Arginina Aldrich X Butilamina Sigma +X X Cadaverina Sigma X Carbonato sódico Panreac, Merck X Carbonato sódico decahidratado Probus X Cisteína Aldrich Cloruro amónico Probus X Cloruro de Cromo (III) Panreac X Cloruro de Cadmio Panreac X +X Cloruro de Cobalto (II) Panreac X X Cloruro de Cobre (II) Panreac X X Cloruro de dansilo Sigma X Cloruro de hexadeciltrimetilamonio Fluka Cloruro de Hierro (III) Panreac X Cloruro de Mercurio (II) Panreac, Merck +X Coproporfirina I dihidrocloruro Aldrich Dicromato potásico Panreac X Dietilamina Sigma X +X X Dimetilamina Sigma +X X Espermidina Sigma X Espermina Sigma X Etilamina Sigma X X Fenilalanina Aldrich Glicocola Aldrich Hexilamina Sigma +X X Hidrógeno carbonato sódico Panreac Hidróxido potásico Probus X
- 59. Capítulo 1. INTRODUCCIÓN 35 Hidróxido sódico Probus, Panreac, Merck X Hipoclorito sódico Prolabo X Histidina Aldrich Imidazol Sigma, Fluka X X Isoleucina Aldrich Isopropilamina Sigma +X X Leucina Aldrich Lisina Aldrich Luminol Fluka X Mercaptoetanol Scharlau X X X Metanol Scharlau X +X Metilamina Sigma X Metionina Aldrich N-acetil-L-cisteina Fluka Nitrato de Co(II) hexahidratado Panreac *Nitrato de cromo (III) Panreac *Nitrato de Niquel hexahidratado Merck X N-Propilamina Sigma +X X Orto-ftaldialdehido Fluka X Óxido de mercurio (II) rojo Panreac +X X Pentilamina Sigma +X X Peróxido de Hidrógeno Panreac, Merck X Polivinil alcohol Probus Prolina Aldrich Putrescina Sigma X Serina Aldrich s-Triazina Fluka Sulfato amónico férrico dodecahidratado Panreac Sulfato amónico ferroso hexahidratado Panreac Sulfato de Cu(II) pentahidratado Merck X X Sulfato de manganeso monohidratado Panreac X Sulfato de Zinc heptahidratado Merck X X Sulfato potásico Prolabo Sulfito sódico Prolabo Tartrato sódico-potásico Panreac Tetraborat o sódico decahidratado Panreac X Tetrahidrofurano Scharlau X +X Tiosulfato sódico Prolabo Tiosulfito sódico Prolabo Tirosina Aldrich X Treonina Aldrich Trietilamina Sigma X Trimetilamina Sigma X Triptófano Aldrich Triton X-100 Fluka X Urea Prolabo Valina Aldrich Yoduro potásico Guinama * Oxidante. Xi: Irritante. Xn: Nocivo. T/T+: Tóxico/muy tóxico. I/I+: Inflamable/Muy inflamable. C: Corrosivo. N: Peligroso para el medio ambiente.
- 60. APARATOS, MONTAJES, INSTRUMENTACIÓN Y SOFTWARE 36 1.5. APARATOS, MONTAJES, INSTRUMENTACIÓN Y SOFTWARE Los aparatos, instrumentos y software utilizados en esta Tesis son los siguientes: § pH-ímetro Crison micropH 2000. § pH-ímetro Crison equipado con un electrodo sensible a gases de amoniaco. § Agitador magnético-Calefactor (Raypa, Spain). § Boma peristáltica Gilson Miliplus 3 (France). § Válvulas para FIA: Rheodyne (Coati, CA) 5041. § Unidad de digestión Kjeldahl: Tecator system 6, 1007 Digestor (Höganas, Sweeden). § Unidad de destilación Kjeldahl: Büchi 323 (Switzerland). § Centrifugadora: Labofugue 200 (Herraeus Sepatch, Germany). § Sistema cromatográfico equipado con una bomba cuaternaria (Hewlett Packard Series 1100, Palo Alto, CA, USA) y una válvula de 6 vías para alta presión Rheodyne modelo 7000 (Figura 7A). Detector UV/vis (Hewlett Packard 1040 M Series II) o detector programable de fluorescencia (Hewlett Packard 1046 A) (Figura 8B). Estaba conectado con el sistema de almacenamiento y procesado de datos Hewlett Packard Chem-Station. Figura 8. A) Equipo cromatográfico HP1100. B)Detectores UV/vis y Fluorímetro. A B
- 61. Capítulo 1. INTRODUCCIÓN 37 § Espectrofotómetro de fila de diodos HP 8452 UV-visible (Germany), equipado con una celda de cuarzo de 1 cm de camino óptico, conectado a un PC Hewlett Packard Vector XM 5/90, controlado por el software G1115AA (Figura 9). Figura 9. Espectrofotómetro HP 8452 conectado a un PC Hewlett Packard Vector XM 5/90. § Sistema cromatográfico equipado con una bomba cuaternaria e inyector automático (Hewlett Packard Series 1050, Palo Alto, CA, USA). Sistema de reacción Post- Columna PCX 5100 (Pickering Laboratories, Mountain View, CA, USA) equipado con dos bombas isocráticas que trabajan a flujo estable. Detector Jasco CL-1525 (Ishika-cho, Hachioji-shi, Tokio, Japan) controlado por el software cromatográfico Borwin y con una interfaz Hercule Lite (JMBS, France) (Figura 10). Figura 10. Equipo cromatográfico 1050, Sistema post-columna PCX-5100, detector Jasco CL-1525 e interfaz Hercule Lite.
- 62. APARATOS, MONTAJES, INSTRUMENTACIÓN Y SOFTWARE 38 § Fluorímetro Hitachi F-4500, 700v (Tokio, Japan) equipado con una celda de 1 cm de camino óptico (Figura 11). Figura 11. Fluorímetro modelo Hitachi F4500. § Fluorímetro Jasco FP-750 (Tokio, Japan), equipado con un módulo agitador y un sistema para la inyección de muestra a través de un septum con una jeringa Hamilton digital (Nevada, USA) (Figura 12). Figura 12. Fluorímetro Jasco FP-750. § Detector quimioluminiscente FireFly CL Detector (Global FIA Inc. P.O. Box 480, Fox Island, WA 98333) (Figura 13), controlado por un registrador o por el software cromatográfico Borwin con una interfaz Hercule Lite (JMBS, France). Figura 13. Detector FireFly CL.
- 63. Capítulo 1. INTRODUCCIÓN 39 Los esquemas utilizados para las separaciones cromatográficas de los derivados dansilados son: § Para la derivatización en línea de aminas alifáticas con Dns-Cl acoplada a HPLC con detección fluorescente (Apéndice 2), el esquema es el que se muestra en la Figura 14: Figura 14. Esquema correspondiente a la derivatización en línea acoplada a la separación cromatográfica (Apéndice 2). § Para la determinación quimioluminiscente de aminas alifáticas o de amonio en agua mediante HPLC (Apéndices 3 y 5 respectivamente), se ha utilizado el mismo esquema (Figura 15) de separación y sistema post-columna acoplado. Figura 15. Esquema correspondiente a la separación cromatográfica de aminas alifáticas (Apéndice 3) / amonio (Apéndice 5) acoplada a un sistema de reacción post- columna con detección quimioluminiscente. Columna Acetonitrilo : Imidazol W Muestra analítica Gradiente elución: Bomba 1 Sistema Post- columna Tampón pH 7 Peróxido de hidrógeno 0.011M Bomba 3 TCPO 2.5 mM Bomba 2 Detector QL 0.35 ml/min 0.35 ml/min Modo de Fluorimetro (Fluorescencia) Bomba 1 Columna Analítica Bomba 2. A W Columna Primaria
- 64. APARATOS, MONTAJES, INSTRUMENTACIÓN Y SOFTWARE 40 En esta Tesis, se han utilizados tres montajes de análisis por inyección en flujo (FIA): § El montaje utilizado en el método propuesto para el screening de muestras de Cu(II) (Apéndice 6), se muestra en la Figura 16: Figura 16. Sistema FIA utilizado en el Apéndice 6. § El montaje utilizado en la elaboración de una guía para la eliminación de los errores sistemáticos en la especiación de Cr(III) y Cr(VI) (Apéndice 7), así como en el estudio de la influencia de los protocolos de acondicionamiento de muestras de agua en la detección quimioluminiscente de elementos traza (Apéndice 8) se muestra en la Figura 17: Figura 17. Sistema FIA utilizado en los Apéndices 7 y 8. Válvula de inyección Luminol 1.2 mM Celda de detección EDTA 0.01M, KOH 0.02 M Muestra Bomba Desecho Instrumento Detector H2O2 0.1 M Bomba 1 v (mL/min) Bomba 2 v (mL/min) Muestra (10-6M Coproporfirina I) CTAC 10-2 M H2O2 0.7 M H2O H2O 0.7 2.2 5 2.9 Baño 100ºC 400 µL 200 µL 200 µL I2 50 µL 50 µL TCPO Detector QL L = 7.5 cm L = 12 cm I1
- 65. Capítulo 1. INTRODUCCIÓN 41 § El montaje utilizado en la determinación del Nitrógeno Orgánico amino y amonio mediante la reacción del luminol con Hipoclorito en medio básico (Apéndice 11) se muestra en la Figura 18: Figura 18. Sistema FIA utilizado en el Apéndice 11. W Celda de detección Bucle: 50 µL I W Reactor, 400 µL Muestra H2O Luminol 1 mM, 0.2M CO3 2- /HCO3 - pH 11.2 Hipoclorito 9 µM, 0.144mM CO3 2- /HCO3 - pH 11.2
- 66. BIBLIOGRAFÍA 42 1.6. BIBLIOGRAFÍA [1] X. Domenech, Química de la Hidrosfera. Origen y destino de los contaminantes, Miraguano, Madrid, 1995. [2] D. Pérez-Bendito, S. Rubio, Environmental Analytical Chemistry, Elsevier, Amsterdam, 1999. [3] I. Bodek, W.J. Lyman, W.F. Reehl, D.H.Roseblatt, Environmental Inorganic Chemistry, Pergamon Press, New York, 1988. [4] M. Radojevic, V.N. Bashkin, Practical Environmental Analysis, RSC, Cambridge, 1999. [5] M.S. Hermann, J.Chem.Ed. 71 (1994) 323. [6] http://www.atsdr.cdc.gov/es/toxfaqs/es_tfacts33.pdf (Agency for toxic substances and disease registry). [7] http://www.atsdr.cdc.gov/es/toxfaqs/es_tfacts132.html (Agency for toxic substances and disease registry). [8] http://www.atsdr.cdc.gov/toxprofiles/tp126-c5.pdf (Agency for toxic substances and disease registry). [9] Y.M. Liu, J.K. Cheng, J. of Chromatogr. A, 1003 (2003) 211. [10] L.A.Tortajada-Genaro, P.Campíns-Falcó, Tecnología del agua, 241 (2003). [11] M.F.Pouet, O.Thomas, B.N.Jacobsen, A.Lynggaard-Jensen, P.Quevauviller, Talanta, 50 (1999) 759. [12] M.Valcárcel, S.Cárdenas, M.Gallego, Trends in Analytcal Chemistry, 18 (11) (1999), 685. [13] M.Valcárcel, S.Cárdenas, M.Gallego, Trends in Analytical Chemistry, 21 (4) (2002), 251. [14] Bruno Enríquez. Energía y tú, 14 (2001). [15] C.Dodeigne, L.Thunus, R.Lejeune. Talanta 51 (2000) 415. [16] H.O. Albrecht, Z. Phys. Chem. 136 (1928) 321. [17] K. Gleu, W. Petsch, Angew Chem. 48 (1935) 57. [18] F. McCapra, P. Richardson, Tetrahedron Lett. (1964) 3167. [19] Encyclopaedia of Analytical Sciences. Volume 5. Academic Press Limited 1995. [20] W.U. Palm, M. Millet, C. Zetzsch, Chemosphere, 35 (5) (1997) 1117. [21] S.W. Tjioe, Robert J. Hurtubise, Talanta, 42 (1) (1995) 59. [22] E. Orejuela, M. Silva, Journal of Chromatogr. A, 1007 (1-2) (2003) 197. [23] Y. Ohba, N. Kuroda, K. Nakashima, Anal. Chim. Acta, 465 (1-2) (2002) 101. [24] C. Molins-Legua, P. Campíns-Falcó, A. Sevillano Cabeza, Anal. Chim. Acta, 378 (1- 3) (1999) 83. [25] K. Honda, J. Sekino, K. Mai, Anal.Chem., 55 (6) (1983) 940. [26] I. Bronstein, B. Edwards, J.C. Voyta, J. Biolumin.Chemilumin., 4 (1) (1989) 99. [27] T. Kinkel, H. Luebbers, E. Schmidt, P. Molz, H. J. Skrzipczyk, J. Biolumin. Chemilumin. 4(1) (1989) 136. [28] P. Jones, T. Williams, L. Ebdon, Anal. Chim. Acta, 237 (1990) 291. [29] K. Tsukagoshi, Y. Obata, R. Nakajima, J. Chromatogr. A, 971 (1-2) (2002) 255. [30] J.Z. Li, P.K. Dasgupta, Anal. Chim. Acta, 442 (1) (2001) 63. [31] T. Pérez-Ruiz, C. Martínez-Lozano, V. Tomás, J. Martín, J. of Chromatogr. A, In Press, Corrected Proof, Available online 4 December 2003.
- 67. Capítulo 1. INTRODUCCIÓN 43 [32] D. Brannegan, M. Ashraf-Khorassani, L.T. Taylor, J-Chromatogr-Sci. 39 (6) (2001) 217. [33] K. Tsukagoshi, Y. Shikata, R. Nakajima, M. Murata, M. Maeda, Anal-Sci., 18 (11) (2002) 1195. [34] E. Pontén, C. Viklund, K. Irgum, S. T. Bogen, Å. N. Lindgren, Anal. Chem., 68 (1996) 4389. [35] X.Yan, J.Chromatogr. A, 842 (1999) 267. [36] A.M. Jiménez, M.J. Navas, Trends in Anal. Chem., 18 (5) (1999) 353. [37] H. Shi, L.T. Taylor, E.M. Fujinari, X. Yan, J. of Chromatogr. A, 779 (1-2) (1997) 307. [38] H. Shi, L.T. Taylor, E.M. Fujinari, J. of Chromatogr. A, 757, (1-2) (1997) 183. [39] D.L. MacTaggart, S.O. Farwell, J.R. Burdge, Z.T. Cai, T.J. Haakenson, W.L. Bamesberger, Atmosph. Environ., 33 (4) (1999) 625. [40] Encyclopedia of Analytical Sciences. Volume 1. Academic Press Limited 1995. [41] R. Escobar, Q.X. Lin, A. Guiraum, F.F. de la Rosa, Analyst, 118 (1993) 643. [42] L.A. Tortajada-Genaro, P. Campíns-Falcó, F. Bosch-Reig, Anal. Chim. Acta, 488 (2) (2003) 243. [43] J. Lu, C. Lau, S. Yagisawa, K. Ohta, M. Kai, J. of Pharmac. and Biomed. Anal., 33 (5) (2003) 1033. [44] Z. Song, C. Wang, Bioorg. & Medic. Chem., 11 (24) (2003) 5375. [45] AM. García-Campaña, W.R.G. Baeyens, X. Zhang, F.Alés, L.Gámiz. Ars Pharmaceutica, 42 (1) (2001) 81. [46] D. Easwaramoorthy, Y. Yu, H. Huang, Anal. Chim. Acta, 439 (1) (2001) 95. [47] W. Qin, Z. Zhang, Y. Peng, Anal. Chim. Acta, 407 (1-2) (2000) 81. [48] J. Li, P.K. Dasgupta, Anal. Chim. Acta, 398 (1) (1999) 33. [49] C. Zhang, J. Huang, Z. Zhang, M. Aizawa, Anal. Chim. Acta, 374 (1) (1998) 105. [50] R. Wei Min, J. Nielsen, J. Villadsen, Anal. Chim. Acta, 312 (2) (1995) 149. [51] S. Ventura, M. Silva, D. Pérez-Bendito, Anal. Chim. Acta, 266 (2) (1992) 301. [52] L.A. Tortajada Genaro, P. Campíns Falcó, S. Meseguer Lloret, F. Bosch Reig, Anal. Bioanal. Chem., 374 (2002) 1223. [53] S. Meseguer Lloret, P. Campíns Falcó, L.A. Tortajada Genaro, F. Blasco Gómez, Inter. J. Environ. Anal. Chem., 83 (5) (2003) 405. [54] Y. Moliner Martínez, S. Meseguer Lloret, L.A. Tortajada Genaro, P. Campins Falcó, Talanta, 60 (2-3) (2003) 257. [55] B. Li, Z. Zhang, Sens. and Act. B: Chemical, 69 (1-2) (2000) 70. [56] Y. Lv, Z. Zhang, F. Chen, Talanta, 59 (3) (2003) 571. [57] H.C. Hong, H.J. Huang, Anal. Chim. Acta, 499 (1-2) (2003) 41. [58] S. Rani Jain, E. Borowska, R. Davidsson, M. Tudorache, E. Pontén, J. Emnéus, Biosens. and Bioelectronics, In Press, Corrected Proof, Available online 23 October 2003. [59] O. Nozaki, H. Kawamoto, Anal. Chim. Acta, 495 (1-2) (2003) 233. [60] G.J. Zhou, G. Wang, J.J. Xu, H.Y. Chen, Sens. and Act. B: Chemical, 81 (2-3) (2002) 334. [61] J.J. Li, J X. Du, J.J. Lu, Talanta, 57 (1) (2002) 53.
- 68. BIBLIOGRAFÍA 44 [62] K.A. Fähnrich, M. Pravda, G.G. Guilbault, Talanta, 54 (4) (2001) 531. [63] H. Lundqvist, C. Dahlgren, Free Radical Biology and Medicine, 20 (6) (1996) 785. [64] D. Kratky, A. Lass, P.M. Abuja, H. Esterbauer, H. Kühn, Biochim. et Biophys. Acta (BBA) - Molecular and Cell Biology of Lipids, 1437 (1) (1999) 13. [65] O.M. Steijger, D.A. Kamminga, A. Brummelhuis, H. Lingeman, J. of Chromatogr. A, 799 (1-2) (1998) 57. [66] J.M. Lin, H. Goto, M. Yamada, J. of Chromatogr. A, 844 (1-2) (1999) 341. [67] W.J. Cooper, J.K. Moegling, R.J. Kieber, J.J. Kiddle, Mar. Chem., 70 (1-3) (2000) 191. [68] C. Shellum, G. Gübitz, Anal. Chim. Acta, 227 (1989) 97. [69] O. Okwumabua, B. Swaminathan, P. Edmonds, J. Wenger, J. Hogan, M. Alden, Research in Microbiology, 143 (2) (1992) 183. [70] K. Matsubara, S. Takeda, Y. Iwaki, J. Cicciarelli, S. Musa, J. Garcia-Gomez, Human Immunology, 44 (1995) 37. [71] M.E. Bolden, N.D. Danielson, J. of Chromatogr. A, 828 (1-2) (1998) 421. [72] B.L. Waguespack, A.Lillquist, J.C. Townley, D.R. Bobbitt, Anal. Chim. Acta, 441 (2) (2001) 231. [73] T. Pérez-Ruiz, C. Martínez-Lozano, V. Tomás, J. Martín, Talanta 58 (5) (2002) 987. [74] N.W. Barnett, T.A. Bowser, R.D. Gerardi, B. Smith, Anal. Chim. Acta, 318 (3) (1996) 309. [75] G.M. Greenway, L.J. Nelstrop, S.N. Port, Analytica Chimica Acta, 405 (1-2) (2000) 43. [76] D.R. Skotty, T.A. Nieman, J. of Chromatogr. B: Biomed. Sci. and Appl., 665 (1) (1995) 27. [77] N.W. Barnett, S.W. Lewis, S.D. Purcell, P. Jones, Anal. Chim. Acta, 458 (2) (2002) 291. [78] S.I. Tu, D. Patterson, J. Uknalis, P. Irwin, Food Research International, 33 (5) (2000) 375. [79] J.I. Horiuchi, K. Ebie, K. Tada, M. Kobayashi, T. Kanno, Bioresource Technology, 86 (1) (2003) 95. [80] B.J. Hindson, N.W. Barnett, Anal. Chim. Acta, 445 (1) (2001) 1. [81] J.W. Costin, P.S. Francis, S.W. Lewis, Anal. Chim. Acta, 480 (1) (2003) 67. [82] Y. Ma, M. Zhou, X. Jin, B. Zhang, H. Chen, N. Guo, Anal. Chim. Acta, 464 (2) (2002) 289. [83] C. Lu, F. Qu, J.M. Lin, M. Yamada, Anal. Chim. Acta, 474 (2002) 107. [84] A.A. Alwarthan, A. Townshend, Anal. Chim. Acta, 205 (1988) 261. [85] M. Matsumoto, Y. Mizoguchi, T. Motoyama, N. Watanabe, Tetrahedron Letters, 42 (50) (2001) 8869. [86] A.I. Voloshin, N.M. Shavaleev, V.P. Kazakov, J. of Luminescence, 91 (1-2) (2000) 49. [87] I. Bronstein, B. Edwards, C. Martin, A. Sparks, J.C. Voyta, Biotechnology Advances, 15 (3-4) (1997) 711. [88] K. Nakashima, M. Wada, N. Kuroda, S. Akiyama, K. Imai, J. Liq. Chromatogr. 17 (10) (1994) 2111.
- 69. Capítulo 1. INTRODUCCIÓN 45 [89] K. Miyaguchi, K. Honda, K. Imai, J. of Chromatogr. A, 303 (1984) 173. [90] J.H. Mike, T.J. Cleland, Anal. Chim. Acta, 259 (1) (1992) 73. [91] M. Cobo, M. Silva, J. of Chromatogr. A, 848 (1-2) (1999) 105. [92] T. Toyo'oka, M. Ishibashi, T. Terao, J. of Chromatography A, 627 (1-2) (1992) 75.
- 70. BIBLIOGRAFÍA 46
- 71. 47 2. RESULTADOS OBTENIDOS Y DISCUSIÓN
- 72. 48
- 73. Capítulo 2. RESULTADOS OBTENIDOS Y DISCUSIÓN 49 2.1. REACCIÓN DEL TCPO 2.1.1. SCREENING DE AMINAS ALIFÁTICAS Y DISMINUCIÓN DE SUS LÍMITES DE DETECCIÓN EN AGUAS. Este apartado se ha desarrollado en tres partes: § Preconcentración y dansilación de aminas alifáticas utilizando cartuchos C18 de extracción en fase sólida y posterior separación cromatográfica de los derivados dansilados con aplicación al análisis de screening en muestras de agua medioambientales. Detección UV o fluorescente. § Automatización del procedimiento anterior: Derivatización en línea de aminas alifáticas con Dns-Cl acoplada a HPLC con detección fluorescente. § Determinación quimioluminiscente de los derivados dansilados de aminas alifáticas formados en cartuchos C18, mediante HPLC. Aplicación a muestras de agua.
- 74. REACCIÓN DEL TCPO 50 2.1.1.1. PRECONCENTRACIÓN Y DANSILACIÓN DE AMINAS ALIFÁTICAS UTILIZANDO CARTUCHOS C18 DE EXTRACCIÓN EN FASE SÓLIDA. APLICACIÓN AL ANÁLISIS DE SCREENING EN MUESTRAS MEDIOAMBIENTALES DE AGUA. Existe muy poca información acerca de la existencia de aminas alifáticas en aguas residuales industriales y en aguas superficiales. La cromatografía gaseosa (GC) [1- 5] y la cromatografía líquida (LC) [6-12] son las principales técnicas utilizadas para la determinación de estos analitos. En la Tabla 10 se muestran algunos de los procedimientos descritos en la bibliografía para la determinación de aminas en muestras de agua a niveles de concentración bajos utilizando GC y LC. Tabla 10. Condiciones de derivatización de aminas alifáticas. Reactivo Muestra Tratamiento de la muestra Detec. LD Ref. Cromatografía Gaseosa (GC) Pentafluobenziladehido Agua residual Deriv. directa 15-30 min a 80 º C, pH 10 y SPME y espaciado de cabeza. FID 0.4- 26 ppb [1] ---- Agua residual SPME y espaciado de cabeza (30min) NPD /MS 3-56 ppb [2] ---- Agua Muestras acidificadas NPD 12-40ppb [3] ---- Agua Muestras acidificadas y espaciado de cabeza (80ºC y 15 min). NPD 0.2-20 ppb 0.01-3ppm [4] Bencenosulfonil cloruro Agua residual y superficial Deriv. directa 30 min a Tª amb. Medio básico y extracción con CH2Cl2. MS 0.1 ppb [5] Cromatografía Líquida (LC) N-hidroxisuccinamidil fluoresceína-O-acetato Patrones Deriv. 30 min 45ºC en tampón borato pH 8.5. FL [2] 3,5-dinitrobenzoil cloruro (DBN) Agua Deriv. en C18-SPE 2 min a Tª amb. En tampón borato pH 10. UV 2-5 ppb [3] Acridina-9-acetil-N- hidroxisuccinimida Agua residual Deriv. directa 10 min a 50ºC en tampón borato pH 8-9. FL 17-87 fmol [4] Cloruro de Dansilo (DNS-Cl) Agua Enriquecimiento en línea en pre - columna IRC-50. Deriv. en línea a 65ºC en tampón borato pH 11. CL 15-300 ppb [5] Fenil isotiocianato Agua de río, grifo, superficial Deriv. directa 15 min a 40ºC en tampón carbonato. Limpieza de la solución derivatizada (C18). UV 0.2-0.6 ppb [6] 4-(5’,6’- dimetoxibenzotizoil) fenilisotiocianato Patrones Deriv. 30 min a 80ºC en NH4OH. FL [7] Fluram Agua Amberlita CG-120, recogida en HCl, evaporada y redisuelta en 1 mL de tampón borato. Deriv. pH 10. UV 0.02-1.2 ppb [8] (FMOC) Agua Deriv. en C18-SPE. 2 min a Tª amb. y pH 10. FL 0.25-5 ppb [13] Cloruro de Dansilo (DNS-Cl) Agua residual Deriv. en C18-SPE 15 min a 85ºC en tampón carbonato pH 9.5. UV- FL 3-15 ppb Este trabajo
- 75. Capítulo 2. RESULTADOS OBTENIDOS Y DISCUSIÓN 51 Debido a las propiedades químicas de las aminas alifáticas, y sus bajos niveles de concentración en las muestras, la mayoría de los procedimientos descritos requieren técnicas de enriquecimiento como la extracción líquido-líquido (LLE), o más recientemente la extracción en fase sólida (SPE), o la micro extracción en fase sólida (SPME), normalmente combinada con derivatización pre- o post-columna en los procedimientos de HPLC. En cromatografía de gases, se ha utilizado la técnica de SPME- espaciado de cabeza [1,2], mejorándose los límites de detección hasta en tres órdenes de magnitud para el análisis de aminas en aire y en matrices acuosas. En HPLC, las aminas de bajo peso molecular normalmente se extraen con resinas Amberlita CG-120 [12] o sorbentes de intercambio catiónico [14], mientras que sus derivados pueden extraerse con sorbentes C18 [10]. M. Cobo y col. [9] han propuesto diferentes estrategias y condiciones combinando derivatización y extracción, que permiten la cuantificación de estos analitos a niveles inferiores a mg/L. La derivatización en disolución ha sido aceptada como un paso efectivo [15], sin embargo, como puede verse en la Tabla 10, existen muchos inconvenientes como el excesivo manejo de la muestra, tiempos de reacción largos, temperaturas elevadas o consumo de disolventes. El uso de SPE para la extracción de los derivados [10] previamente formados en disolución, puede resolver algunos problemas, y gracias a los elevados factores de concentración, se pueden obtener límites de detección similares o incluso menores a los obtenidos utilizando GC-MS con una instrumentación mucho más sencilla [5]. Sin embargo, los problemas relacionados con el proceso de derivatización todavía permanecen (manejo de la muestra, falta de automatización, etc.). Una solución posible a los problemas de pasos extra y de interferencias, es llevar a cabo la etapa de derivatización en el soporte sólido [16]. En esta dirección, este grupo de investigación ha desarrollado una metodología en la que todos los pasos de la preparación de la muestra (extracción, concentración, derivatización y transferencia al sistema cromatográfico) se llevan a cabo en el mismo soporte, en línea o fuera de línea. El método se basa en atrapar los analitos en el sorbente, purificarlos con un disolvente adecuado y derivatizarlos pasando el reactivo a través de los cartuchos. El analito y el reactivo se dejan reaccionar durante un período de tiempo, y después el exceso de reactivo se elimina (si se requiere) pasando un disolvente adecuado a través de los cartuchos. Por último, los derivados, se desorben y se recogen para procesarlos posteriormente [17] o son transferirdos a la columna primaria en un sistema en línea [18-19]. Se ha demostrado recientemente la utilidad de esta metodología utilizando el reactivo-UV 3,5-dinitrobenzoil cloruro (DNB) y las aminas alifáticas (etilamina, isopropilamina y dimetilamina) [7]. También se ha estudiado la derivatización asistida en soporte sólido de algunas aminas alifáticas con 9-fluorenilmetil cloroformiato (FMOC) [13]. Esta metodología la han aplicado recientemente D. Shangguan y col. [20] en la determinación de aminoácidos y péptidos con FMOC y sorbente de sílice. En este trabajo, se aplica la mencionada metodología al análisis de screening de alquilaminas en muestras de agua utilizando el reactivo Cloruro de Dansilo (Dns-Cl) como reactivo derivatizante. Las derivatizaciones con DnsCl se llevan a cabo
- 76. REACCIÓN DEL TCPO 52 habitualmente en medio acuoso-acetona saturado con carbonato sódico en tiempos de reacción largos y a elevadas temperaturas de reacción, y en muchos casos, se requiere la extracción de los analitos derivatizados para eliminar el reactivo en exceso. Sin embargo, es interesante el estudio de procedimientos de dansilación simplificados debido a los límites de detección tan bajos que se alcanzan. Estos límites de detección pueden mejorarse porque los derivados dansilados proporcionan quimioluminiscencia con el ácido oxálico bis(2,4,6-tricloro fenil éster) (TCPO/H2O2). Se propone un método rápido y sencillo para la preconcentración y derivatización seguido de su separación mediante HPLC y detección de aminas en muestras de agua medioambientales a niveles de µg/l. Se estudian sistemáticamente 7 aminas alifáticas y amonio, para obtener las mejores condiciones analíticas de retención y dansilación en cartuchos C18. Se optimizan las condiciones de separación cromatográfica. Los derivados de dansilo se registran a 333 nm con detección UV, y a λexc = 350 nm / λem = 530 nm con detección fluorescente. Finalmente, el método se aplica a la determinación o screening de las aminas estudiadas en muestras de agua. Más información acerca de las condiciones experimentales empleadas, se refleja en el Apéndice 1 [21]. Derivatización en disolución La respuesta obtenida para las diferentes aminas al derivatizarlas en disolución, se tomó como referencia para las medidas en cartucho (conversión del 100%). El exceso de reactivo se destruyó al calentar, por lo que no interfirió al tiempo de retención de los analitos. Preconcentración y derivatización en cartuchos C18 Los cartuchos Bond Elut C18, se seleccionaron para la preconcentración y purificación de las aminas alifáticas basándonos en estudios previos [17]. No se observó elución al pasar 1 mL de muestra ni al pasar el reactivo (Dns-Cl 12.5 mM en acetona : tampón carbonato 33.3 mM pH 9.5 (2:3, v/v)) a través del cartucho. Se estudiaron los parámetros de la reacción de derivatización: pH del medio, tiempo y temperatura de reacción. Los intervalos estudiados para cada parámetro manteniendo fijos los otros dos, así como los óptimos seleccionados, se muestran en la Tabla 11. Al estudiar el pH de la reacción se observó que el rendimiento de la reacción aumentaba hasta pH 9.5, a partir del cual se mantuvo constante (Figura 19 A). El estudio del tiempo de reacción, proporcionó rendimientos similares entre 15 y 45 minutos. Un tiempo de reacción de 60 minutos se consideró excesivo (Figura 19 B). El estudio de la Temperatura de reacción, reveló un aumento importante del rendimiento al utilizar 85 ºC, que se mantuvo hasta 110ºC (Figura 19 C). No se observaron productos de degradación en las condiciones óptimas seleccionadas (pH 9.5, Tª 85ºC, Tiempo 15 min).
- 77. Capítulo 2. RESULTADOS OBTENIDOS Y DISCUSIÓN 53 Tabla 11. Parámetros de reacción estudiados. Parámetro Intervalo Parámetros fijos Óptimo Seleccionado pH 8.5-11.4 25ºC, 30 min pH 9.5 Temperatura 25-100 ºC pH 9.5, 15 min 85 ºC Tiempo 0-60 min pH 9.5, 85 ºC 15 min Al utilizar detección UV, la determinación de hexilamina presentó dificultades debido a su coelución junto con el exceso de reactivo, por lo que se requirió un paso de lavado del exceso de reactivo previo a la desorción del soporte de los derivados dansilados formados. Para ello se utilizó una disolución saturada de carbonato en presencia de hidróxido sódico. Se optimizó la concentración de NaOH en el intervalo 0.1- 0.3 M, seleccionando como óptima la concentración 0.1 M, pues a concentraciones superiores el rendimiento de la reacción disminuía entre un 20 y un 4% en función de la amina estudiada. Por otra parte, al utilizar detección fluorescente, no hubo problemas de interferencia con el exceso de reactivo, lo que simplificó el procedimiento en el cartucho eliminando la fase de lavado del reactivo. Como disolvente de elución de los derivados dansilados del soporte, se utilizó 1 mL de acetonitrilo. 40 60 80 100 120 140 160 180 200 8 9 10 11 12 pH Areadepico A) 0 100 200 300 400 500 600 700 800 0 20 40 60 Tiempo (min) AreadePico B) 0 100 200 300 400 500 600 20 40 60 80 100 Temperatura (ºC) AreadePico C) Figura 19. Área de Pico frente a: A) pH de la reacción. B) Tiempo de reacción. C) Temperatura de la reacción. ♦ Metilamina, Etilamina, • N- Propilamina, ×× Butilamina, ∗ Pentilamina. Condiciones: DnsCl 5 mM; Tampón borato 20 mM; concentración de amina 5 mg/l.
- 78. REACCIÓN DEL TCPO 54 Con estos parámetros optimizados, se evaluó la eficiencia del método de derivatización asistida en soporte sólido propuesto, comparando las áreas de pico obtenidas utilizando el procedimiento en cartucho con las obtenidas en disolución, y se concluyó que el procedimiento en cartucho era más efectivo que en disolución. En la Tabla 12 se puede ver como todas las recuperaciones son superiores al 100%. Esto se debe a que el reactivo y la amina se concentran en el soporte sólido facilitando la reacción. Las desviaciones estándar relativas también mejoran al utilizar la derivatización en cartucho. Tabla 12. Comparación entre la derivatización en cartucho y en disolución. Derivatización en disolución Derivatización SPE C18 Analito DER (%) %REC DER (%) NH4 + 25 (320 ± 70) 22 Metilamina 25 (123 ± 9) 7 Etilamina 26 (135 ± 14) 10 N-Propilamina 20 (146 ± 16) 11 Butilamina 15 (130 ± 16) 12 Dietilamina 17 (160 ± 20) 13 Pentilamina 15 (133 ± 11) 8 Hexilamina 14 (163 ± 19) 12 El volumen de muestra a procesar, se estudió desde 1 a 25 mL con la intención de aumentar el factor de preconcentración, de forma que la concentración final fuera la misma en todos los ensayos. Los porcentajes de recuperación de la señal obtenidos tomando como referencia la señal para 1 mL de muestra procesado, para cada amina en los diferentes volúmenes estudiados, se reflejan en la Tabla 13. Tabla 13. Porcentajes de Recuperación en función del Volumen de muestra procesado. Volumen Muestra (mL) 1 5 15 25 50 100 Analito Porcentaje de Recuperación (%) NH4 + 100 87.8 39.3 29.4 MA 100 79.0 50.3 37.4 26.1 9.4 EA 100 81.1 44.6 31.6 19.1 8.7 DMA 100 92.5 63.2 73.8 BA 100 82.5 52.6 48.5 37.5 15.7 DEA 100 58.3 42.3 34.6 PeA 100 93.7 63.5 61.5 11.6 11.8 HA 100 88.5 58.8 63.4 35.1 17.5
- 79. Capítulo 2. RESULTADOS OBTENIDOS Y DISCUSIÓN 55 A mayores volúmenes, se observaron pérdidas por elución, mayores cuanto mayor era el carácter polar del analito. Como compromiso, se seleccionaron 5 mL para los ensayos posteriores. En cuanto al volumen de disolvente utilizado para la desorción de los productos de reacción, se utilizó la mínima cantidad posible para alcanzar el máximo factor de preconcentración, obteniéndose porcentajes de recuperación similares al utilizar 0.5 o 1 mL de acetonitrilo. Se intentó mejorar la retención de las aminas alcalinizando los patrones a pH 11, y las recuperaciones fueron entre el 85% y el 115% respecto a los patrones neutros. Sin embargo, para el amonio se obtuvo una recuperación del 40%, debido a su transformación a amoniaco. Separación cromatográfica Los derivados dansilados se separaron en el sistema cromatográfico estando el orden de elución relacionado con su carácter polar. Los tiempos de retención fueron: 2.2, 3.2, 3.9, 4.8, 5.7, 6.2, 6.7, 7.8 y 8.2 para NH4 + , MA, EA, DMA, BA, PeA, HA y 1,7- diaminoheptano respectivamente. En la Figura 20, se muestran los cromatogramas típicos para un blanco y una mezcla de aminas (10 mg/l NH4 + , 7.5 mg/l DEA y 2.5 mg/l del resto de aminas) para un volumen procesado de 1 mL y condiciones de reacción óptimas, con detección UV (Figura 20 A) o de fluorescencia (Figura 20 B)). En 9 minutos el cromatograma se completa para los 7 analitos y el patrón interno. En fluorescencia, el blanco fue más limpio y el ruido de fondo menor. Características analíticas En la Tabla 14, se muestran las curvas de calibrado obtenidas para cada amina utilizando detección UV y fluorescente, así como los intervalos lineales y límites de detección. La sensibilidad varió en función de la amina estudiada, y los límites de detección mejores fueron del orden de µg/l con ambos tipos de detección al procesar 5 mL de muestra. El amonio pudo identificarse, pero no cuantificarse. Los estudios de precisión dieron medidas de repetibilidad (en un mismo día) algo mejores que las medidas de reproducibilidad (entre días). Las medidas de repetibilidad para diferentes volúmenes de muestra estudiados se muestran en la Tabla 15, siendo mejores a menor volumen de muestra procesado. Los valores de desviación estándar relativa para las medidas de reproducibilidad, se situaron entre el 7 y el 15 %.
- 80. REACCIÓN DEL TCPO 56 Figura 20. Cromatogramas para blanco ( ) y mezcla de aminas ( ) con detección A) UV a 333 nm, y B) Fluorescencia λexc = 350 nm / λem = 530 nm. 0 1 2 3 4 5 6 7 8 9 0 10 20 30 40 50 Absorbancia Tiempo (min) NH4+ MA DMA BA EA PeA HA DEA A) 0 1 2 3 4 5 6 7 8 9 0 10 20 30 40 50 Fluorescencia Tiempo (min) NH4+ MA DMA BA EA PeA HA DEA B)
- 81. Capítulo 2. RESULTADOS OBTENIDOS Y DISCUSIÓN 57 Tabla 14. Características analíticas para los derivados amina-dansilo con detección UV o fluorescente. Analito (a ±± sa) (b ±± sb) sy/x n r 2 Intervalo lineal (µµg/l) LD (µµg/l) D MA (7 ± 3) (470 ± 10) 6.3 15 0.991 10-500 2 FL EA (1 ± 2) (465 ± 9) 4 9 0.997 10-500 2 FL DMA (10 ± 2) (255 ± 7) 3 10 0.996 10-500 3 FL BA (-1 ± 1) (346 ± 7) 3 15 0.995 10-500 3 FL DEA (8 ± 1) (74 ± 2) 2 7 0.990 30-1500 4 FL PeA (-4 ± 2) (425 ± 10) 6 15 0.990 10-500 2 FL HA (-3 ± 2) (388 ± 10 ) 6 14 0.992 10-500 2 FL MA (18 ± 4) (567 ± 19) 8 15 0.992 10-500 3 UV EA (2 ± 3) (495 ± 18) 8 14 0.990 10-500 6 UV DMA (64 ± 4) (442 ± 17) 10 8 0.990 10-500 6 UV BA (-1.6 ± 2.5) (331 ± 14) 7 14 0.990 10-500 9 UV DEA (5 ± 2) (66± 3) 3.5 11 0.990 30-1500 15 UV PeA (-2.9 ± 4) (306 ± 18) 7 11 0.990 10-500 8 UV HA (10 ± 1) (177 ± 4) 2 12 0.997 20-500 15 UV MA* (-1 ± 6) (85 ± 3) 10 6 0.996 100-5000 48 UV EA* (-11 ± 4) (58 ± 2) 7 8 0.997 100-5000 93 UV DMA* (-2 ± 2) (47.5 ± 0.8) 3 10 0.9992 100-5000 75 UV BA* (-18 ± 4) (36 ± 1) 5 8 0.995 100-5000 90 UV DEA* (11 ± 5) (7 ± 1) 4 4 0.990 350-15000 340 UV PeA* (-11 ± 4) (37 ± 2) 6 9 0.991 100-5000 90 UV HA* (-20 ± 4) (35 ± 1.5) 5 6 0.994 100-5000 70 UV Condiciones: Volumen procesado: 5 mL (o 1 mL)*; Volumen de elución: 0.5 mL (o 1 mL)*. Tabla 15. Estudios de precisión. Repetibilidad (Desviación Estándar Relativa %). Repetibilidad DER( %) Analito 1 mL 5 mL 10 mL 25 mL MA 8 4 4 4 EA 4 4 9 14 N-para 4 - 8 15 BA 4 8 6 2 DEA(*) - 14 15 8 PeA 4 - 6 3 HA - 4 3 - Concentración: 5 mg/L para 1 mL. *15 mg/L para 1 mL.
- 82. REACCIÓN DEL TCPO 58 Análisis de muestras de agua Para validar la exactitud del método se fortificaron cuatro muestras de agua reales (Muestra 1: agua de riego, Muestra 2: agua de lago, Muestra 3: agua de grifo, Muestra 4: agua residual) con cantidades conocidas de los analitos (Método de Adición Estándar), utilizando patrón interno para mejorar los resultados. Las pendientes obtenidas para los analitos más polares (amonio, metilamina, etilamina y dimetilamina) aplicando el MOSA, fueron menores que las obtenidas con patrones, lo que indicaba la presencia de efecto matriz. Sin embargo, para analitos no polares (dietilamina, butilamina y pentilamina), el comportamiento al aplicar el MOSA fue similar al obtenido con patrones. Además se observó que este comportamiento en las recuperaciones, no dependía de la concentración, lo que se refleja en la Figura 21. Figura 21. Porcentajes de recuperación en dos muestras de agua diferentes para las diferentes aminas a diferentes niveles de concentración. Los porcentajes de recuperación para las tres muestras de agua procesadas utilizando 5 mL de muestra (Muestras 1, 2, y 3), se reflejan en la Tabla 16. Se puede deducir un porcentaje de recuperación medio para cada analito puesto que no hubo diferencias en las recuperaciones entre las diferentes muestras procesadas. Para deducir la concentración en muestras, habría que aplicar el método de adición estándar o bien utilizar la curva de patrones corregida por el factor de recuperación correspondiente a cada analito. 0 0.2 0.4 0.6 0.8 1 1.2 1.4 0 0.2 0.4 0.6 Conc (ppm) %Rec(Alturadepico) NH MA EA DMA BA PEA DEA 0 0.2 0.4 0.6 0.8 1 1.2 1.4 0 0.1 0.2 0.3 Conc (ppm) %Rec(Alturadepico) NH MA EA DMA BA PEA DEA
- 83. Capítulo 2. RESULTADOS OBTENIDOS Y DISCUSIÓN 59 Tabla 16. Porcentajes de recuperación y desviación estándar relativa para cada analito en las diferentes muestras de agua. Muestra 1 Muestra 2 Muestra 3 %Rec Medio Analito % Rec DER (%) % Rec DER (%) % Rec DER (%) % Rec DER (%) NH4 + 45±3 6 25±3 14 38±6 17 37±9 25 MA 46±9 19 38±2 5 65±9 14 51±14 27 EA 69±7 11 79±5 7 88±19 22 80±14 17 DMA 74±5 6 74±3 4 83±13 16 78±9 11 BA 101±17 17 112±4 3 103±20 19 106±13 12 DEA 109±11 10 105±4 4 109±14 13 107±8 8 PeA 94±18 20 112±4 3 119±14 11 108±16 15 La muestra de agua residual analizada, Muestra 4, contuvo una concentración de aminas elevada, por lo que se requirió menor volumen para su análisis (0.2 mL de muestra se llevaron a 1 mL). El screening de esta muestra (Figura 22) mostró la presencia de amonio, metilamina, dimetilamina y pentilamina. Figura 22. Cromatogramas para: a) Muestra 4. b) Muestra 4 fortificada con la mezcla de 1.25 mg/L de cada amina. La fortificación de esta muestra con una mezcla de todas las aminas, dio recuperaciones del orden del 100% para todas las aminas, lo que indicaba la ausencia de efecto matriz. Las pendientes del MOSA fueron comparadas con un test t de comparación de pendientes, y resultaron similares a las pendientes de las curvas de calibrado de patrones con una probabilidad del 99%. 0 2 4 6 8 10 Absorbancia Tiempo (min) NH4+ EA DEA BA PeA HA I.S. NH4+ MA DMA MA DMA PeA I.S. a) b)
- 84. REACCIÓN DEL TCPO 60 Para esta muestra, las concentraciones deducidas para cada analito utilizando el MOSA o la curva de calibrado con patrones se muestran en la Tabla 17. Como puede observarse, los errores relativos son aceptables en todos los casos. Tabla 17. Concentraciones deducidas en la muestra de agua residual (% Error relativo) Concentraciones deducidas Analito Concentrac. Adicicionada (mg/l) MOSA Calibrado patrones 1.5 1.14 (8.8%) 1.13 (9.6%) Metilamina 2.5 2.61 (4.4%) 2.56 (2.4%) 1.5 1.34 (7.2%) 0.98 (21.6%) Etilamina 2.5 2.49 (0.4%) 2.58 (3.2%) 1.5 1.13 (9.6%) 1.18 (5.6%) Dimetilamina 2.5 2.56 (2.4%) 2.40 (4%) 1.5 1.22 (2.4%) 1.08 (13.6%) Butilamina 2.5 2.52 (0.8%) 2.56 (2.4%) 1.5 1.08 (13.6%) 1.21 (3.2%) Pentilamina 2.5 2.57 (2.8%) 2.50 (0%) 3.75 3.75 (0%) --- Dietilamina 7.5 7.50 (0%) 7.51 (0.13%) Conclusiones La derivatización en cartuchos C18, fue más efectiva que la derivatización en disolución. Las condiciones óptimas de derivatización fueron: pH de la reacción 9.5, temperatura y tiempo de reacción: 85 o 100ºC y 15 o 10 minutos respectivamente, y volumen de muestra a procesar 5mL o 1 mL. El procedimiento propuesto fue simple y rápido y consumió menos tiempo que otros procedimientos de dansilación, siendo el tiempo total de análisis (tratamiento de la muestra más cromatografía) menor de 25 minutos. Se pudieron procesar hasta 5 mL de muestra con pocas pérdidas en las aminas más polares. Las recuperaciones en muestras fueron independientes del tipo de muestra analizada, pero dependientes del analito. El efecto matriz pudo corregirse aplicando el MOSA o utilizando la curva de calibrado de patrones teniendo en cuenta los porcentajes de recuperación. En muestras cuyo contenido en aminas es a niveles de mg/L, como la muestra de agua residual, el volumen a procesar fue de 0.2 mL diluido a 1 mL. En este caso, no hubo efecto matriz, siendo las recuperaciones cercanas al 100% para todas las aminas.
- 85. Capítulo 2. RESULTADOS OBTENIDOS Y DISCUSIÓN 61 El procedimiento propuesto pudo aplicarse con exactitud y reproducibilidad satisfactorias a la determinación de siete aminas alifáticas a niveles de concentraciones inferiores a los mg/L, y también para la identificación de amonio. El procedimiento se utilizó para el screening de amonio y aminas en una muestra de agua residual.
- 86. REACCIÓN DEL TCPO 62 2.1.1.2. DERIVATIZACIÓN EN LÍNEA DE AMINAS ALIFÁTICAS CON Dns -Cl ACOPLADA A HPLC CON DETECCIÓN FLUORESCENTE. La marcada tendencia en Química Analítica hacia el desarrollo de sistemas completamente automatizados, requería alguna aportación en este sentido. Los procedimientos de trabajo en línea, se basan en los mismos principios que los procedimientos de trabajo fuera de línea. La instrumentación requerida así como el manejo de la misma, es mínimo frente al equipamiento convencional. Normalmente, no requieren la continua intervención del operador, lo que se traduce en una menor variabilidad y menores errores sistemáticos. Con esta intención, y continuando con el trabajo anterior, se propuso la derivatización en línea de aminas alifáticas con cloruro de dansilo acoplada a HPLC con detección fluorescente. Condiciones de enriquecimiento, lavado y derivatización Para la reacción de derivatización en línea de aminas alifáticas con cloruro de dansilo acoplada a la separación cromatográfica de los derivados dansilados formados con detección fluorescente, se utilizó el montaje mostrado en la Figura 14 del Capítulo 1-Introducción (Pág. 39). En este montaje, las muestras se inyectaban en una precolumna empaquetada con material de relleno C18 para el enriquecimiento, lavado y derivatización de la muestra con el reactivo Dns-Cl. Más información acerca de las condiciones experimentales se refleja en el Apéndice 2 [22]. Se optimizaron las condiciones de retención en la precolumna así como de derivatización, tales como: volumen de muestra, concentración del reactivo, tiempo de reacción, temperatura de reacción, relación reactivo:tampón y características de la fase móvil. En la Tabla 18, se muestran los intervalos estudiados y las condiciones óptimas escogidas en función de la máxima retención de los analitos en la precolumna, así como la máxima reactividad en cuanto a la formación del derivado dansilado. Tabla 18. Condiciones optimizadas e intervalos estudiados. Parámetro Intervalo Óptimo Concentración DnsCl 2.5 mM - 12.5 mM 5 mM Volumen muestra 150 - 400 µL 150 µL Temperatura 20 - 80ºC 60ºC Tiempo reacción 10 - 30 min 10 min Relación Reactivo:tampón 40:60 o 30:70 40:60 Fase móvil Tampón pH 12 o H2O H2O
- 87. Capítulo 2. RESULTADOS OBTENIDOS Y DISCUSIÓN 63 Condiciones cromatográficas Además de las condiciones de retención y derivatización, se establecieron las condiciones cromatográficas óptimas para el análisis de screening de aminas alifáticas en muestras de agua utilizando cloruro de dansilo como reactivo de derivatización. Estas condiciones así como la programación de tiempos se muestra en la Tabla 19. Tabla 19. Condiciones cromatográficas experimentales para la retención, derivatización, separación y detección en línea de aminas alifáticas. Tiempo (min) Precolumna Columna Analítica Posición de la válvula 0 0-3.75 9-13.5 Inyección de Muestra:150µL Inyección de Dns-Cl 5 mM: 40 µL Inyección de tampón: 60 µL Lavado de la muestra: Gradiente de 0.5 mL/min a 0 en 3.75 min 50:50(v/v)(acetonitrilo:imidazol pH 7) (v flujo = 0.1 mL/min) 50:50(v/v)(acetonitrilo:imidazol pH 7) (gradiente v flujo = 0.1 a 1 mL/min) A 13.5-16.5 Separación Analítica 70:30 (v/v) Acetonitrilo:Imidazol pH 7 50:50(v/v)(acetonitrilo:imidazol pH 7) modo gradiente (v flujo = 1.0 mL/min) B 16.5-28.5 28.5-29.5 30.5 H2O:Acetonitrilo(gradiente a 100% acetonitrilo) H2O:Acetonitrilo (gradiente a 100% agua) Fin 50:50(v/v)(acetonitrilo:imidazol pH 7) modo gradiente hasta 90% MeCN (v flujo = 1.0 mL/min) 50:50(v/v)(acetonitrilo:imidazol pH 7) (gradiente v flujo = 1 a 0.1 mL/min) A Características analíticas Las curvas de calibrado lineales obtenidas para metilamina, etilamina, dimetilamina, butilamina, dietilamina, pentilamina y hexilamina utilizando detección fluorescente, así como los intervalos dinámicos de concentración, tiempos de retención (tr), límites de detección (calculados como la concentración correspondiente a una relación señal/ruido de 3) y otras figuras de mérito, se muestran en la Tabla 20. La sensibilidad varió en función de la amina estudiada, obteniendo mayor sensibilidad y menores límites de detección al aumentar la longitud de la cadena alifática. Los cromatogramas típicos en las condiciones de reacción optimizadas, para un blanco y una mezcla de aminas de concentración 1.25 mg/l para MA, EA y DMA, 0.75 mg/l para BA, 3.75 mg/l para DEA y 0.375 para PeA y HA, se muestran en la Figura 23. Como puede observarse, la separación de las 7 aminas es buena y después de la derivatización, el registro del cromatograma completo solo requiere 9 minutos.
- 88. REACCIÓN DEL TCPO 64 Tabla 20. Curvas de calibrado, parámetros analíticos y tiempos de retención para la derivatización en línea de aminas alifáticas con Dns-Cl. Amina (a ±± sa) (b ±± sb) IL (mg/L) n r 2 sy/x LD (ìg/L) tr (min) MA (-40 ± 60) (130 ± 40) 0.5-2.25 3 0.921 60 7 1.86 EA (-50 ± 40) (90 ± 30) 0.5-2.25 3 0.922 30 15 2.4 DMA (52 ± 10) (289 ± 10) 0.5-2.5 5 0.996 19 11 3.07 BA (5 ± 13) (420 ± 30) 0.075-1.35 3 0.996 14 0.6 4.24 DEA (42 ± 16) (32.8 ± 1.4) 0.15-22.5 5 0.995 20 3 4.81 PeA (-30 ± 120) (2900 ± 200) 0.015-1 10 0.951 200 0.03 5.85 HA (-70 ± 50) (3360 ± 50) 0.015-2.5 11 0.998 120 0.05 7.28 Figura 23. Cromatogramas para la derivatización en línea de aminas alifáticas con Cloruro de dansilo para un blanco y una mezcla de aminas. Los estudios de precisión proporcionaron valores de repetibilidad buenos en las condiciones óptimas estudiadas, siendo los porcentajes de desviación estándar relativa (%DER) 19.8, 11.7, 10.8, 19.6, 18.4, 6.3 y 3.4 % para MA, EA, DMA, BA, DEA, PeA y HA respectivamente (Concentraciones: 1.5 mg/l para MA, EA y DEA; 0.5 mg/l para DMA, BA y PeA; y 0.2 mg/l para HA). 0 2 4 6 8 10 40 80 120 160 200 MA EA DMA BA DEA PeA HA S.I. Tiempo (min) Fluorescencia Blanco Mezcla de aminas
- 89. Capítulo 2. RESULTADOS OBTENIDOS Y DISCUSIÓN 65 Análisis de muestras de agua Se analizó mediante este procedimiento la muestra de agua residual, que se fortificó con la mezcla de aminas. Los porcentajes de recuperación obtenidos para las diferentes aminas: 17%, 66%, 13%, 21%, 14%, 73% y 102% para MA, EA, DMA, BA, DEA, PeA y HA respectivamente, aumentaron al aumentar la longitud de la cadena alifática. Esta presencia de efecto matriz, habría de corregirse utilizando el método de adición estándar si la finalidad del análisis fuera la determinación. Basándonos en estos resultados, concluimos que podemos utilizar el procedimiento propuesto para el screening de aminas alifáticas en aguas. El cromatograma obtenido para la muestra de agua residual (Figura 24), mostró la presencia de compuestos como metilamina, dimetilamina y pentilamina en esta muestra. Figura 24. Cromatograma de screening de una muestra de agua residual. Conclusiones Las condiciones óptimas de retención en la precolumna y derivatización fueron: 150 ìL de muestra, DnsCl 5 mM, Tª 60ºC, 10 minutos de reacción, relación reactivo: tampón 40:60, y fase móvil agua. Con la optimización de las condiciones 0 2 4 6 8 10 40 80 120 160 200 MA DMA PeA HA I.S. Fluorescencia Tiempo (min)
- 90. REACCIÓN DEL TCPO 66 cromatográficas, en 30 minutos, se consiguió en línea la retención, derivatización y separación cromatográfica de las 7 aminas alifáticas. Este sistema presentó la ventaja de la completa automatización frente al sistema presentado en el capítulo anterior (Apéndice 1, [21]). Además, las condiciones de la reacción de derivatización fueron algo menos drásticas, con la ventaja de que la misma precolumna podía reutilizarse inyección tras inyección. En el caso de la derivatización en cartuchos de extracción en fase sólida, un mismo cartucho solamente podía reutilizarse una sola vez. Se consiguió mejorar los límites de detección para Butilamina, Pentilamina y Hexilamina, aunque para metilamina, etilamina, dimetilamina y dietilamina, los límites de detección alcanzados fueron peores. Los estudios de precisión dieron %DER inferior al 20% en todas las aminas. La sensibilidad varió en función de la amina estudiada, aumentando al aumentar la longitud de la cadena alifática. Los estudios de recuperación de la cantidad fortificada en la muestra, dieron un comportamiento similar al obtenido al utilizar el procedimiento fuera de línea (Apéndice 1, [21]), aumentando los porcentajes de recuperación al aumentar la longitud de la cadena alifática. El efecto matriz habría de corregirse utilizando el MOSA para las aminas de cadena corta. El procedimiento es aplicable al screening de aminas en muestras de agua.
- 91. Capítulo 2. RESULTADOS OBTENIDOS Y DISCUSIÓN 67 2.1.1.3. DETERMINACIÓN DE AMINAS ALIFÁTICAS EN AGUA MEDIANTE HPLC CON DETECCIÓN QUIMIOLUMINISCENTE. Las aminas alifáticas son compuestos que se encuentran a bajas concentraciones en algunos tipos de aguas, por lo que los métodos para su análisis han de ser suficientemente sensibles. La sensibilidad y la selectividad necesarias se pueden alcanzar por combinación de la derivatización de los analitos con técnicas de separación (cromatografía líquida (LC), cromatografía gaseosa (GC) o electroforesis capilar (CE)) y un método de detección de elevada sensibilidad (métodos quimioluminiscentes (QL), electroquímicos o espectrometría de Masas). La técnica de detección quimioluminiscente ha demostrado ser una herramienta sensible para la medida de niveles ultra trazas, con sistemas ópticos baratos y simples que proporcionan bajo ruido de fondo y un amplio intervalo dinámico [23]. La quimioluminiscencia de compuestos sintéticos como la lofina, el Luminol, los ésteres de acridinio, ésteres de perioxalato y complejos de rutenio, se ha utilizado ampliamente. Derivados del Luminol como el N-(4-(aminobutil)N-etilisoluminol (ABEI), NN’dissinimidilcarbonato (DSC) [24-25], 4-isotiocianatoftalhidracina (ILITC) [26], 6- isotiocianatobenzo(g)ftalacina-1, 4(2H,3H)-diona (IPO) [27], y ácido 4-(6,7-dihidro-5,8- dioxotiazolo (4,5-g)ftalacina-2-il) benzoico N-hidroxisuccinimida éster (TPB-suc) [28], se han utilizado como reactivos quimioluminiscentes para la determinación de compuestos amino. El Tris(bipiridil)rutenio(III) [Ru(bipi)3] se ha empleado para la determinación de una gran variedad de compuestos de nitrógeno sin necesidad de derivatizar el analito [24,29]. El método quimioluminiscente del [Ru(bipi)3] es muy selectivo para la determinación de aminas terciarias, pero no muy sensible comparado con otras técnicas de determinación quimioluminiscente. Este reactivo normalmente se electrogenera, dando lugar a quimioluminiscencia electrogenerada (ECL) [30]. A pesar del elevado potencial teórico de la ECL, solamente ciertas reacciones han encontrado aplicación en análisis, debido a limitaciones como la presencia de especies interferentes, su compleja optimización y su falta de reproducibilidad [31]. En presencia de un fluoróforo, los derivados oxálicos son los emisores quimioluminiscentes no biológicos más eficientes. Los más utilizados recientemente son el Bis-(2,4,6.triclorofenil)oxalato (TCPO), el bis (2,4-dinitrofenil)oxalato (DNPO) y el Bis[2-(3,6,9-trioxadeciloxicarbonilfenil]oxalato. Las principales ventajas de estos sistemas son la eficiencia y la flexibilidad. Estos sistemas requieren la presencia de un compuesto fluorescente, obtenido normalmente a través de la reacción de derivatización del analito. Se han utilizado reactivos derivatizantes como el Cloruro de Dansilo (Dns-Cl) [32-33,18] para aminas biogénicas y anfetaminas, el naftaleno-2,3-dialdehido (ADA) [34] para aminas primarias, y la luminarina [35] para aminas primarias y secundarias, combinados con mezclado postcolumna con los reactivos TCPO y H2O2. En este trabajo, se estudia la generación de quimioluminiscencia post-columna con el sistema TCPO/H2O2, para aumentar la sensibilidad del procedimiento de dansilación previamente expuesto (Apéndice 1, [21]) para aminas de bajo peso molecular
- 92. REACCIÓN DEL TCPO 68 en muestras de agua medioambientales, alcanzándose niveles por debajo de los ng/mL. Se estudian sistemáticamente seis aminas alifáticas: metilamina, etilamina, butilamina, pentilamina, hexilamina y dietilamina. Además, se incluyen también aminas biogénicas como putrescina, cadaverina, espermina y espermidina en el estudio de screening. Por último, se aplica el método a la determinación de aminas en muestras de agua reales. Más información acerca de las condiciones experimentales se refleja en el Apéndice 3 [36]. Los resultados obtenidos con detección quimioluminiscente, se comparan con los obtenidos con detección UV y fluorescente utilizando el reactivo cloruro de dansilo [21, 22] u otros reactivos como cloruro de 3,5-dinitrobenzoilo (DNB) [7], fluorenilmetilcloroformiato (FMOC) [13] y orto-ftaldialdehido/N-Acetil-L-Cisteína (OPA/NAC) [37]. Condiciones de preconcentración y condiciones pre y post columna Se utilizaron las condiciones de preconcentración y derivatización establecidas previamente en el Apéndice 1 (Pág. 52-55). Los productos de reacción formados se eluyeron con 0.5 mL o 1 mL de acetonitrilo. Los derivados dansilados se inyectaron en el sistema cromatográfico y se mezclaron después de su separación, con el TCPO y el H2O2. En cuanto a las condiciones postcolumna, los parámetros que afectaban a la respuesta quimioluminiscente de acuerdo con los estudios previos [38-39] eran: temperatura, pH, cantidad de agua, catalizador, disolvente, concentración de TCPO, concentración de H2O2, volumen del tubo de mezclado, volumen de la celda de detección y velocidades de flujo de los diferentes canales. Los parámetros establecidos para este trabajo fueron: temperatura ambiente, acetonitrilo como disolvente, imidazol como catalizador, velocidades de flujo inferiores a 0.6 mL/min, longitud del tubo de mezclado mínima para el montaje, volumen de la celda de detección fijado por el instrumento de medida, concentración de TCPO superior a 0.1 mM y relación TCPO:H2O2 cercana a 5. Los parámetros más críticos, la concentración y el pH del Imidazol (evaluados en la bibliografía en los rangos 0.1-5 mM y pH 5-7), dieron la máxima relación señal/ruido a concentración 1 mM y pH 7, siendo la velocidad del flujo 1.5 mL/min. El producto de reacción entre el TCPO y el H2O2 (el 2,4,6-triclorofenol), puede dar lugar a efecto quenching disminuyendo la señal quimioluminiscente. Para evitar este producto, ambos reactivos se prepararon por separado y se mezclaron en el sistema (ver montaje en la Figura 15 del Capítulo 1-Introducción, Pág. 39). Se observó que la sensibilidad era 5 veces superior cuando el TCPO (preparado en una mezcla acetonitrilo:tetrahidrofurano), se mezclaba con los derivados antes que el H2O2. La concentración de TCPO se varió entre 0.5-5mM a 0.35 mL/min, aumentando la sensibilidad al aumentar la concentración del TCPO, pero no se observaron diferencias entre las concentraciones 2.5 mM y 5 mM, por lo que se eligió como concentración óptima de TCPO 2.5 mM. Al variar la concentración del H2O2, se observó que la intensidad de QL disminuía al aumentar la concentración por encima de 5 mM, además
- 93. Capítulo 2. RESULTADOS OBTENIDOS Y DISCUSIÓN 69 de aumentar la línea base (ruido de fondo), mientras que al trabajar en el intervalo 11- 5mM, la señal se mantuvo estable. En la Tabla 21, se resumen las condiciones experimentales óptimas para los pasos pre y post columna. Tabla 21. Condiciones pre y postcolumna óptimas para la determinación de aminas. Paso Parámetros experimentales Pre-columna: Preconcentración Derivatización en cartuchos C18 1 mL Metanol 1 mL tampón carbonato pH 12, 10mM 1 mL/5 mL muestra 0.5 ml reactivo Dns-Cl Tª 110ºC-10 min Elución 1 mL Acetonitrilo Separación Cromatográfica de los derivados dansilados. Inyección de muestra: 20 µl Gradiente: 0 min. 50:50, 9 min (80:20) (MeCN:imidazol pH 7), v flujo = 1.5 mL/min Post-columna: Reacción Quimioluminiscente de los derivados dansilados previamente separados. TCPO 2.5 mM, 0.35 ml/min H2O2 11 mM, 0.35 ml/min La eficiencia de la reacción para cada amina medida por separado, respecto a la medida en la mezcla de las seis aminas, se evaluó mediante los porcentajes de recuperación de señal (Señal mezcla*100/Señal por separado), los cuales fueron: (108±14), (100±11), (114±3), (88±3), (78±5) y (109±16) para MA, EA, BA, PeA, HA y DEA respectivamente. Estos porcentajes tan próximos al 100%, indicaron que no había diferencias significativas al trabajar con la mezcla de aminas. Para aumentar el factor de enriquecimiento se estudió el volumen de muestra a procesar entre 1 y 25 mL. En la Figura 25, puede observarse como al utilizar 25 mL, se produjeron importantes pérdidas por elución de los analitos, de modo que para mejorar los límites de detección en la determinación de las aminas, podían utilizarse volúmenes de 5 o 10 mL. 0 20 40 60 80 100 %Recuperación MA EA BA DEA PeA HA V = 1 ml V = 5 ml V = 10 ml V = 25 ml Figura 25. Porcentajes de recuperación para las diferentes aminas estudiadas en función del volumen de muestra procesado.
- 94. REACCIÓN DEL TCPO 70 Condiciones cromatográficas Para la separación de las seis aminas alifáticas, se empleó el Gradiente de Fase Móvil 1 (acetonitrilo:imidazol 50:50 a t = 0 min, 80:20 a t = 9 min, 50:50 a t = 10min), y los cromatogramas típicos obtenidos muestran la total separación de las seis aminas (Figura 26) en tan solo 7 minutos, con tiempos de retención 2.27, 2.79, 4.33, 4.81, 5.27 y 6.31 min, para MA, EA, BA, DEA, PeA y HA respectivamente. En esta misma figura, se muestra también el cromatograma del blanco yel cromatograma correspondiente a una mezcla que incluye además de estas 6 aminas, 4 aminas biogénicas (putrescina, cadaverina, espermina y espermidina). Como puede observarse en la Figura 26, los picos correspondientes al blanco no interfirieron en los picos de las aminas en estudio. También se observó que utilizando este gradiente de elución, la Putrescina coeluye con la Pentilamina. 0 4 8 12 16 Tiempo (min) SeñalQuimioluminiscente Fase Móvil 1 Blanco Aminas Alifáticas Aminas Alifáticas + Aminas Biogénicas R R R R R R 1 2 3 4 5 6 1 2 34 5+7 8 6 9 10 Figura 26. Cromatogramas correspondientes a blanco, patrón de aminas alifáticas y patrón de aminas alifáticas + aminas biogénicas, utilizando el gradiente de Fase Móvil 1. R- Reactivo, 1-MA (concentración 0.15 mg/l), 2- EA (0.240 mg/l), 3-BA (0.3 mg/l), 4- DEA (0.48 mg/l), 5-PeA (0.3 mg/l), 6-HA (0.3 mg/l), 7-Put (0.3 mg/l), 8-Cad (0.3 mg/l), 9-Spd (0.3 mg/l), 10-Spm (1.12 mg/l). El Gradiente de Fase Móvil 2 (acetonitrilo:imidazol 50:50 isocrático de a t = 0 a 13 min, gradiente hasta 90:10 a t = 17 min, 50:50 a t = 20min), se empleó para la separación de las 10 aminas (alifáticas + biogénicas). El cromatograma completo del blanco y de una mezcla de las 10 aminas se muestra en la Figura 27. El registro con las
- 95. Capítulo 2. RESULTADOS OBTENIDOS Y DISCUSIÓN 71 10 aminas separadas duró 20 minutos y los tiempos de retención fueron: 2.41, 3.17, 6.38, 7.54, 9.51, 10.74, 13.65, 14.54, 17.6 y 18.52 minutos, para MA, EA, BA, DEA, PeA, Put, Cad, HA, Spd y Spm respectivamente. Este gradiente permitió la separación de los picos de Pentilamina y Putrescina que coeluían al utilizar el Gradiente de Fase móvil 1. 0 4 8 12 16 20 Tiempo (min) SeñalQuimioluminiscente R R R R 1 2 3 4 5 7 8 6 9 10 Fase Móvil 2 Aminas Alifáticas + Aminas Biogénicas Blanco Figura 27. Cromatogramas correspondientes a blanco y patrón de aminas alifáticas + aminas biogénicas, utilizando el gradiente de Fase Móvil 2. R-Reactivo, 1-MA (concentración 0.15 mg/l), 2- EA (0.240 mg/l), 3-BA (0.3 mg/l), 4- DEA (0.48 mg/l), 5-PeA (0.3 mg/l), 6-HA (0.3 mg/l), 7-Put (0.3 mg/l), 8-Cad (0.3 mg/l), 9-Spd (0.3 mg/l), 10-Spm (1.12 mg/l). Curvas de calibrado y parámetros analíticos Para evaluar las posibilidades cuantitativas del método propuesto, se estudiaron las curvas de calibrado Altura de Pico frente a Concentración (mg/l) para cada amina utilizando la Fase Móvil 1. En la Tabla 22, se muestran los resultados obtenidos utilizando volúmenes de patrón de 1, 5 y 10 mL. Se observa buena linealidad en todos los casos, estando los intervalos lineales en los µg/L. Las sensibilidades (pendiente de los calibrados) aumentaron del orden de 4-5 veces al procesar volúmenes de 5 mL frente a 1 mL, y del orden de 10 veces al procesar volúmenes de 10 mL frente a 1 mL. Los límites de detección establecidos como la concentración requerida para generar una relación señal/ruido de 3, para los volúmenes de patrón procesados de 1, 5
- 96. REACCIÓN DEL TCPO 72 (elución con 0.5 o 1 mL de acetonitrilo), 10 y 25 mL, se muestran en la Tabla 23. Los mejores límites de detección se alcanzaron procesando 5 mL de patrón y eluyendo los derivados dansilados con 0.5 mL de acetonitrilo, o bien procesando 10 mL de muestra y eluyendo con 1 mL de acetonitrilo. Tabla 22. Parámetros analíticos de los derivados amina-dansilo detectados por quimioluminiscencia. Amina V (mL) b ±± sb (L· mg -1 ) a ±± sa (Voltios) sy/x r 2 n LI (µµg/L) MA 1 1900 ± 120 25 ± 14 17 0.9887 5 17-200 EA 1 1010 ± 30 -3 ± 5 6 0.9987 4 3-320 BA 1 570 ± 40 -8 ± 9 12 0.9904 4 13-400 PeA 1 820 ± 50 -8 ± 12 15 0.9920 4 7-400 HA 1 630 ± 20 2 ± 6 7 0.9969 4 7-400 DEA 1 239 ± 17 7 ± 6 8 0.9856 5 13-640 MA 5 10500 ± 500 16 ± 14 15 0.9973 3 4.3-40 EA 5 4300 ± 500 -17 ± 18 20 0.9629 5 0.7-64 BA 5 1790 ± 160 -5 ± 8 9 0.9838 4 3-80 PeA 5 3000 ± 300 -9 ± 14 16 0.9732 5 2-80 HA 5 2800 ± 200 -9 ± 10 11 0.9847 5 1.7-80 DEA 5 860 ± 50 3 ± 4 5 0.9919 4 4-128 MA 10 22300 ± 1500 52 ± 10 15 0.9915 4 3-10 EA 10 10100 ± 500 3 ± 6 9 0.9944 4 0.5-16 BA 10 5600 ± 600 4 ± 8 12 0.9787 4 2-20 PeA 10 9000 ± 500 1 ± 7 10 0.9935 4 1-20 HA 10 10300 ± 300 5 ± 4 6 0.9984 4 1-20 DEA 10 2870 ± 150 2 ± 3 5 0.9943 4 3-32 Tabla 23. Limites de detección (µg/L) obtenidos con detección quimioluminiscente a diferentes volúmenes de muestra y diferentes volúmenes de elución. Comparación con los valores obtenidos con otros sistemas de detección y reactivos. Detección Reactivo QL DNSCl UV DNSCl FL DNSCl UV DNB FL FMOC FL OPA/NAC V Muestra V Elución 1 ml 1 ml 5 ml 1 ml 5 ml 0.5 ml 10 ml 1 ml 25ml 1 ml 5 ml 0.5 ml 5 ml 0.5 ml 5 ml 0.5 ml 5 ml 0.5 ml 10 ml 2 ml MA 5 1.3 0.9 0.8 2 3 2 - 0.5 5 EA 0.8 0.2 0.15 0.15 0.2 6 2 5 0.25 6 BA 4 0.9 0.5 0.6 0.8 9 3 - 1 10 PeA 2 0.6 0.3 0.3 0.5 8 2 - 5 23 HA 2 0.5 0.3 0.3 0.3 15 2 - 2.5 - DEA 4 1.3 0.8 0.8 0.9 15 4 - - -
- 97. Capítulo 2. RESULTADOS OBTENIDOS Y DISCUSIÓN 73 Los estudios de precisión en función del volumen de patrón procesado se muestran en la Tabla 24. Los valores de desviación estándar relativa fueron adecuados independientemente del volumen de muestra, siendo los valores de repetibilidad similares a los de reproducibilidad. Se seleccionó como volumen óptimo 5 mL. Si bien la precisión fue algo pobre para la dietilamina, para otras aminas como la pentilamina, se mejoró este parámetro de un 20 a un 0.9% (ver Tabla 24). Tabla 24. Estudios de precisión (DER %). Repetibilidad y reproducibilidad del método utilizando diferentes volúmenes de muestra. Repetibilidad (% DER) Reproducibilidad (% DER)Analito 1 ml 1 ml 5 ml 10 ml Metilamina 6 0.8 11 7 Etilamina 10 4 10 17 Butilamina 3 6 16 13 Pentilamina 5 20 0.9 9 Hexilamina 11 1.3 3 8 Dietilamina 16 10 29 9 Análisis de muestras de agua El método propuesto se aplicó al análisis de varias muestras de agua. La fortificación de las muestras con cantidades conocidas de los analitos, se utilizó para validar la exactitud del método. En la Tabla 25 se muestran las concentraciones fortificadas encontradas para cada una de las muestras analizadas. Para las muestras de agua de grifo (S1), agua de riego (S2), agua de lago (S3), y agua residual (S4), las concentraciones halladas por el método fueron muy cercanas a las reales adicionadas, siendo los porcentajes de recuperación medios (± desviación estándar): 113 (± 22, n=7), 100 (± 19, n=4), 114 (± 11, n=7) y 99 (± 11, n=12) % para S1, S2, S3 y S4 respectivamente. Estos valores se consideraron aceptables teniendo en cuenta los niveles de concentración ensayados. En todos los casos, indicaron la ausencia de efecto matriz. Sin embargo, la muestra de agua marina analizada (S5), dio porcentajes de recuperación cercanos al 100% (101 ± 22) para aminas con 1C o 2C, mientras que aminas con mayor número de átomos de C, dieron porcentajes de recuperación elevados, entre 150 y 200 %. Por tanto, para las aminas con más de 2 C, debido al efecto matriz, la concentración en esta muestra debería calcularse utilizando el método de adición estándar (MOSA) en lugar de la curva de calibrado de patrones.
- 98. REACCIÓN DEL TCPO 74 Tabla 25. Concentración de amina fortificada y concentración de amina fortificada encontrada (µg/L) en muestras de agua reales. Concentración fortificada (µg/L) Concentración encontrada (µg/L) Muestra MA EA BA PeA HA DEA MA EA BA PeA HA DEA 20 32 40 64 15 31 50 88 S1 80 80 128 106 97 136 S2 20 32 40 40 19 26 39 51 40 40 47 51 S3 40 64 80 80 128 52 72 80 88 133 100 160 200 200 200 320 90 120 178 194 186 360 100 200 200 200 112 222 189 202S4 200 200 218 211 20 32 17 39 S5 40 65 80 31 68 110 Se encontró y cuantificó Metilamina utilizando la curva de calibrado de patrones en agua de lago (8.8±0.3 µg/l), agua de riego (7±3 µg/l) y agua residual (6000±700 µg/l), en la que también se encontró Pentilamina (3100±200 µg/l). En agua de grifo, se encontró y cuantificó Dietilamina utilizando la curva de calibrado de patrones (13±3, n=3) µg/l y utilizando la curva de adición estándar (15 µg/L, n=5), siendo los resultados similares. En agua de mar no se detectaron ninguna de las aminas estudiadas. Las muestras de agua residual y de agua marina, se analizaron también utilizando el procedimiento cromatográfico descrito en el Capítulo previo (Apéndice 1, [21]), utilizando dansilación asistida en soporte sólido y detección fluorescente. Con este procedimiento, no se detectó ninguna amina al analizar la muestra de agua marina, al igual que en el método propuesto, y para la muestra de agua residual los resultados fueron (6040 ± 900 µg/L, n=3) y (2960 ± 160 µg/L, n=3) para metilamina y pentilamina respectivamente. Ambos valores pueden considerarse estadísticamente iguales (con un nivel de confianza del 95%) a los obtenidos por el método propuesto. El cromatograma obtenido para el análisis de screening de la muestra de agua residual (Figura 28) utilizando la Fase Móvil 1, indicó la presencia de metilamina y putrescina y/o pentilamina; utilizando la Fase Móvil 2, se confirmó la presencia de pentilamina y metilamina. Los límites de detección obtenidos utilizando el procedimiento de screening propuesto para las aminas alifáticas y biogénicas en el agua residual, calculado como 3 veces la señal del fondo dividido por la pendiente, fueron: 0.066, 0.053, 0.015, 0.2, 0.011, 0.0075, 0.027, 0.0076, 0.011 y 0.06 mg/L para MA, EA, BA, DEA, PeA, Put, Cad, HA, Spd y Spm respectivamente.
- 99. Capítulo 2. RESULTADOS OBTENIDOS Y DISCUSIÓN 75 0 4 8 12 16 20 Tiempo (min) SeñalQuimioluminiscente R 1 R R 1 R 5 o 7 5 Fase M óvil 1 Fase M óvil 2 Figura 28. Cromatogramas correspondientes a una muestra de agua residual utilizando la Fase Móvil 1 y la fase Móvil 2. R-Reactivo, 1-MA, 5-PeA y 7-Put. Comparación con otros sistemas de detección La detección quimioluminiscente permitió alcanzar los límites de detección más bajos, como se observa en la Tabla 23. Para cada procedimiento se han considerado los mejores volúmenes de muestra y de elución. Utilizando los derivados dansilados, los límites de detección pueden mejorar desde 75 a 3 veces respecto a las medidas con detección UV, y desde 10 a 2 veces respecto a las medidas en fluorescencia dependiendo de la amina. Por otra parte, otros reactivos fluorescentes como FMOC y OPA/NAC, dieron límites de detección más elevados que los que proporcionó la detección quimioluminiscente. Los valores de repetibilidad y reproducibilidad de la detección fluorescente y quimioluminiscente utilizando derivados dansilados, fueron comparables. Para la detección fluorescente, se obtuvieron %DER entre 4 y 14% (calculadas de 3 réplicas de 5 mL de mezcla de aminas a concentraciones de 1mg/L excepto DEA a 3 mg/L). Los valores para la detección QL son los mostrados en la Tabla 24. En el trabajo de dansilación de aminas con detección UV o fluorescente, se requirió un paso de limpieza en el soporte sólido para eliminar el blanco reactivo. Este paso producía una elución parcial de algunas aminas, especialmente de las más polares, lo que se observó también al aplicar el procedimiento a muestras reales, requiriéndose la
- 100. REACCIÓN DEL TCPO 76 aplicación del MOSA para calcular las concentraciones en las muestras. Sin embargo, al utilizar la detección quimioluminiscente, este paso de lavado no se incluyó en el procedimiento, obteniéndose recuperaciones más elevadas para las aminas más polares, con lo que no apareció efecto matriz. La única excepción fue la muestra de agua marina que, para el análisis de aminas con más de dos átomos de C, requirió la aplicación del MOSA. Conclusiones En esta parte de la Tesis, se estudió la detección quimioluminiscente de aminas para mejorar sus límites de detección. Para ello, la dansilación se realizó en soporte sólido como tratamiento precolumna. Los derivados dansilados se inyectaron en el sistema cromatográfico y se mezclaron con TCPO/H2O2 para generar la quimioluminiscencia. Se consiguió la cuantificación de las aminas alifáticas a niveles de ìg/L con exactitud y reproducibilidad satisfactorias. Estos niveles de concentración son adecuados para la monitorización de las aminas alifáticas en muestras de agua reales. No hubo diferencias significativas en la cuantificación de los analitos entre los diferentes tipos de muestra ensayados. El método también se empleó con propósitos de screening en presencia de otras aminas alifáticas como las poliaminas espermina, espermidina, putrescina y cadaverina. Se optimizó un procedimiento de screening para separar las diez aminas.
- 101. Capítulo 2. RESULTADOS OBTENIDOS Y DISCUSIÓN 77 2.1. DISMINUCIÓN DEL LÍMITE DE DETECCIÓN DE AMONIO EN AGUA E INCREMENTO DE LA EXACTITUD DE SU DETERMINACIÓN. El amonio es un micronutriente en los sistemas acuosos. Tiene un papel fundamental en el ciclo del nitrógeno. Cuando se encuentra en concentraciones elevadas, la cantidad de nutrientes aumenta en los sistemas acuosos aumentando la actividad biológica, que lleva a un incremento del crecimiento de algas, turbidez, olor, mal sabor y toxicidad. El amonio puede encontrarse en aguas superficiales, subterráneas o marinas a bajas concentraciones, del orden de 10 µg/L. En aguas residuales, puede hallarse a elevadas concentraciones, del orden de 30 mg/L debido a los procesos de amonificación y de reducción de nitrato. La legislación especifica las características del método de análisis que se utilice para estimar su concentración. Su exactitud, precisión, y límite de detección han de ser el 10% del valor paramétrico [40]. Los métodos de referencia como la detección espectrofotométrica de amonio utilizando el reactivo de Nessler, la reacción del indofenol o el electrodo selectivo de amonio, se han utilizado ampliamente [41]. Además, en los últimos años se han desarrollado nuevos métodos. En la Tabla 26, se resumen algunas de las características analíticas como el límite de detección, el intervalo dinámico lineal y la técnica empleada, para algunos de los métodos publicados en la bibliografía. Los métodos de inyección en flujo han sido los más destacados en los últimos años, normalmente combinados con sistemas como colectores de flujo [47], detectores de fluorescencia con guía de ondas [49], sistemas quimioluminiscentes [50], detección fluorimétrica basada en la complejación de amonio con o-ftaldialdehido y tioglicolato [53], y detector de fila de diodos con el reactivo de Nessler [44]. También se han descrito métodos que utilizan cromatografía iónica [46,48] y electrodos selectivos de amonio [42,43,51,54].
- 102. REACCIÓN DEL TCPO 78 Tabla 26. Resumen de algunos procedimientos descritos en la bibliografía para la determinación de amonio. Ref. Técnica LD (µµg/L) Intervalo Lineal (mg/L) [42] Método de lavado para la determinación en flujo de amonio con un electrodo iónico de gases. - 0.1 – 10 [43] Determinación NH4 + en flujo. Detector electrodo de gases. 3.85 3.857e-3 – 1.2857 [44] Sistema FIA con 3 detectores: potenciométrico, conductimétrico y fotométrico (DAP). Reactivo de Nessler. - 0.05 - 0.9 [45] Sistema FIA con pre-concentración en resina de intercambio catiónico. Basado en la reacción de Nessler. 3 0.05 - 0.5 [46] Cromatografía iónica con detección fila de diodos. 18 0.36– 18 [47] Sistema FIA con colector de flujo. Detección fluorimétrica basada en la reacción OPA/sulfito/NH4 + y pre-concentración 0.054 9e-5 - 0.135 [48] Cromatografía iónica. 12.8 0.05 – 5 [49] Sistema FIA con detección fluorimétrica utilizando una guía de ondas, basado en la reacción OPA/sulfito/NH4 + . 1.87 0.0107 - 0.0535 [50] Sistema FIA quimioluminiscente basado en la reacción del luminol. 7.2 0.0535 – 5.35 [51] Biosensor iónico amperométrico basado en un electrodo de carbono modificado. 107 0.107-1.3375 [52] Sistema FIA con detección amperométrica indirecta de los iones amonio. Electrodo modificado de Clinoptilolito. 90 0.36-18 [53] Sistema FIA con detección fluorimétrica. Basado en la reacción OPA/tioglicolato. 2.675e-3 – 5.35 [54] Electrodo selectivo de amonio. 0.155 0.18-1.8 El estudio de la determinación de amonio se ha abordado en dos partes, con la intención de aportar nuevos métodos que mejorasen la sensibilidad y selectividad de los de referencia: § En la primera parte, se desarrolla un método fluorimétrico para la determinación de amonio en muestras de agua basado en la formación de un isoindol. Además se hace un estudio comparativo con los métodos de Nessler y Electrodo Selectivo de amonio, que se revisan previamente a su utilización. § En la segunda parte, se propone un método para la determinación quimioluminiscente de amonio en aguas por HPLC. El amonio se derivatiza en disolución con el reactivo cloruro de dansilo, y posteriormente el derivado se separa mediante HPLC y se hace confluir post-columna con TCPO y H2O2 generando la señal quimioluminiscente.
- 103. Capítulo 2. RESULTADOS OBTENIDOS Y DISCUSIÓN 79 2.1.2.1. DETERMINACIÓN DE AMONIO EN MUESTRAS DE AGUA UTILIZANDO EL REACTIVO OPA-NAC. ESTUDIO COMPARATIVO CON LOS MÉTODOS DE NESSLER Y ELECTRODO SELECTIVO DE AMONIO. En este capítulo, se propone un método basado en la derivatización de amonio con el reactivo o-Ftaldialdehido (OPA) y N-Acetil-L-Cisteína (NAC) en condiciones básicas. El derivado isoindólico formado se detectó fluorimétricamente a λexcitación = 415 nm y λemisión = 485 nm o λexcitación = 333 nm y λemisión = 462.5 nm. Se establecieron las condiciones óptimas para obtener la máxima sensibilidad y selectividad, y los resultados se compararon con los obtenidos utilizando el método del reactivo de Nessler y el método del electrodo selectivo. Los tres métodos ensayados, se examinaron quimiométricamente para evaluar la presencia o ausencia de errores sistemáticos y mejorar sus incertidumbres al analizar muestras desconocidas. Se compararon las ventajas y desventajas de cada uno de ellos en términos de sensibilidad, selectividad, exactitud y precisión. Más información acerca de las condiciones experimentales se refleja en el Apéndice 4 [55]. Método de OPA-NAC Se observó una variación de la señal fluorescente del isoindol formado en la reacción amonio/OPA/NAC con el tiempo de reacción, diferente en función de la longitud de onda seleccionada. A λexc = 415 nm / λemisión = 485 nm, la señal aumentaba con el tiempo, alcanzando un máximo a 5 minutos (óptimo seleccionado) (Figura 29 a); por otra parte, a λexc = 333 nm / λem = 462.5 nm, la señal disminuía con el tiempo, seleccionándose como óptimo 2 minutos (Figura 29 b). Los espectros de emisión fluorescente para un blanco y un patrón de amonio, a λexc = 415 nm / λemisión = 485 nm (t reacción 5 min) y λexc = 333 nm / λem = 462.5 nm (t reacción 2 minutos), se muestran en la Figura 30 a) y b) respectivamente. Se optimizaron las condiciones de la reacción de derivatización: concentraciones del reactivo OPA/NAC y pH del Tampón Borato, utilizando un diseño factorial 32 (Tabla 27) a dos niveles de pH (10.6 (-) y 10.8 (+)), dos concentraciones de OPA (8.8 mM (-) y 15 mM (+)), y dos concentraciones de NAC (8.8 mM (-) y 60 mM (+)). Las condiciones óptimas (máxima sensibilidad) seleccionadas fueron: 8.8 mM OPA:NAC (1:1) a pH 10.8. En la Tabla 28 se dan los resultados en cuanto a la importancia e interacción entre los factores estudiados siendo el valor Dreferencia = 14.88.
- 104. REACCIÓN DEL TCPO 80 Figura 29. Señal fluorescente frente al tiempo. a) λexc = 415 nm / λem = 485 nm; b) λexc = 333 nm / λem = 462.5 nm. Figura 30. Espectros de emisión de fluorescencia. a) λexc = 415 nm / λem = 485 nm (t reacción 5 min); b) λexc = 333 nm / λem = 462.5 nm (t reacción 2 min). Tabla 27. Diseño factorial de optimización de las concentración de reactivos y pH. pH OPA NAC 1 + - - 2 + + - 3 + + + 4 + - + 5 - - - 6 - + - 7 - + + 8 - - + 0 300 600 900 1200 1500 0 5 10 15 Tiempo (min)Fluorescencia b) NH4+ Blanco 0 300 600 900 1200 1500 0 5 10 15 Tiempo (min) Fluorescencia a) Blanco NH4+ 0 500 1000 1500 2000 2500 420 470 520 570 λλ emisión (nm) Fluorescencia a) Blanco 1.4 mg/L NH4+ 0 500 1000 1500 2000 2500 380 480 580 λλ emisión (nm) Fluorescencia b) Blanco 1.4 mg/L NH4+
- 105. Capítulo 2. RESULTADOS OBTENIDOS Y DISCUSIÓN 81 Tabla 28. Importancia e interacción entre los factores: pH, [OPA] y [NAC]. Factor o interacción Valor D Conclusión PH 32.82 DpH > Dreferencia OPA 58.65 DOPA > Dreferencia NAC 976.50 DNAC > Dreferencia Importancia de los factores: [NAC] > [OPA] > pH PH/OPA 11.26 DpH/OPA < Dreferencia No hay interacción PH/NAC 32.17 DpH/NAC > Dreferencia OPA/NAC 58.34 DOPA/NAC > Dreferencia Dos interacciones importantes: OPA/NAC > pH/NAC Las curvas de calibrado de amonio a las diferentes λexc y diferentes tiempos de reacción, y otros parámetros analíticos como límites de detección, límites de cuantificación y desviación estándar del procedimiento, se muestran en la Tabla 29. Como la reactividad del OPA-NAC con aminas primarias es bien conocida, se estudió su interferencia en la determinación de amonio a las dos longitudes de onda de excitación. A λexc = 415 nm, MA, EA, IPA y β-FEA no dieron ninguna señal fluorescente significativa, incluso a concentraciones del orden de 15 y 30 mg/l. La curva de calibrado de amonio en presencia de 1 mg/L de MA (Tabla 29) corrobora este hecho, manteniendo tanto la pendiente como la ordenada igual a la curva de calibrado de patrones sin MA. Por otra parte, en la Tabla 29 se observa que a λexc = 333 nm, la pendiente de la curva de calibrado en presencia de MA, fue similar a la obtenida con patrones sin MA; no así ocurrió para la ordenada, lo que indica que a esta λexc, la metilamina está contribuyendo a la señal obtenida. Tabla 29. Curvas de calibrado de amonio y parámetros analíticos utilizando el reactivo OPA-NAC. Condiciones t (min) a ±± sa b ±± sb (n, r 2 , sy/x) DL (mg/L) QL (mg/L) SDP (sy/x/b) OPA/NAC 8.8mM (1:1), λexc = 415 nm 5 48 ± 11 759 ± 7 (10, 0.9983, 17) 0.07 0.2 0.02 OPA/NAC 8.8mM (1:1), λexc = 333 nm 5 610 ± 30 360 ± 40 (6, 0.943, 60) 0.5 1.6 0.16 OPA/NAC 8.8mM (1:1), λexc = 333 nm 2 450 ± 50 1380 ± 70 (6, 0.9899, 90) 0.19 0.6 0.06 * OPA/NAC 8.8mM (1:1), λexc = 415 nm 5 60 ± 11 737 ± 16 (5, 0.9985, 18) 0.07 0.2 0.02 * OPA/NAC 8.8mM (1:1), λexc = 333 nm 2 1380 ± 110 1380 ± 160 (5, 0.962, 180) - - 0.13 * Calibrado de amonio en presencia de 1 mg/l de MA.
- 106. REACCIÓN DEL TCPO 82 Estudios de confirmación: Método de OPA-NAC, Nessler y Electrodo Selectivo. Revisión del Método de Nessler En primer lugar, se estudió la influencia de la preparación del reactivo Nessler (en exceso de HgCl2 (NR1) o con la cantidad justa hasta la aparición del precipitado rojo (NR2)). Los resultados obtenidos fueron similares requiriéndose únicamente en el caso del NR2 la adición de 100 µl de NaOH 6N. Se varió el volumen de reactivo Nessler utilizado para la reacción entre 50-300 µl (en 2.5 ml), seleccionándose como óptimo 100 µl. Como agente complejante de interferentes, se estudió la adición de EDTA o de Tartrato sódico-potásico, pero el EDTA impidió la reacción, por lo que se seleccionó tartrato de concentración 0.708 mM. Para eliminar el precipitado que se formaba a elevadas concentraciones de amonio (superiores a 2.5 mg/l) se utilizó PVA y Triton X- 100, pero ninguno de ellos consiguió eliminarlo. En la Tabla 30 se muestran las curvas de calibrado de amonio y otros parámetros analíticos a diferentes tiempos de reacción y en diferentes condiciones experimentales. Puede observarse que las pendientes, los límites de detección y cuantificación y la desviación estándar del procedimiento son similares en todas las condiciones estudiadas. Tabla 30. Curvas de calibrado de amonio y parámetros analíticos utilizando el Método de Nessler. Condiciones t (s) ( b ±± sb ) ( a ±± sa ) sy/x , r2 , n LD mg/L LC mg/L DEP sy/x/b 100 µl NR1, Sin tartrato 600 (0.126 ± 0.003) (0.041 ± 0.006) 0.013, 0.997, 9 0.3 1.1 0.11 0 (0.123 ± 0.003) (0.044 ± 0.008) 0.014, 0.997, 8 0.4 1.2 0.12 210 (0.123 ± 0.003) (0.049 ± 0.008) 0.015, 0.997, 8 0.4 1.2 0.12 390 (0.123 ± 0.003) (0.052 ± 0.007) 0.014, 0.997, 8 0.3 1.1 0.11 100 µl NR1, 0.0708 M tartrato 600 (0.123 ± 0.003) (0.054 ± 0.007) 0.013, 0.997, 8 0.3 1.1 0.11 0 0.130 ± 0.007 0.096 ± 0.009 0.014, 0.992, 5 0.3 1.1 0.11100 µl NR1, 0.708 mM tartrato. H2O nanopura tratada 120 0.125 ± 0.006 0.105 ± 0.008 0.012, 0.994, 5 0.3 1.0 0.10 50 µl NR1, 0.708 mM tartrato 600 (0.1018 ± 0.0011) (0.0266 ± 0.0017) 0.003, 0.999, 4 0.08 0.3 0.03 0 (0.125 ± 0.003) (0.010 ± 0.009) 0.014, 0.998, 5 0.3 1.2 0.12 210 (0.131 ± 0.006) (0.018 ± 0.015) 0.02, 0.995, 5 0.6 1.8 0.18 390 (0.132 ± 0.006) (0.018 ± 0.015) 0.02, 0.994, 5 0.6 1.9 0.19 100 µl NR2, 100 µl NaOH, 0.708 mM tartrato 600 (0.132 ± 0.06) (0.018 ± 0.015) 0.02, 0.994, 5 0.6 1.9 0.19 0 0.1196 ± 0.0017 0.011 ± 0.005 0.007, 0.999, 5 0.18 0.6 0.06 210 0.125 ± 0.005 0.020 ± 0.012 0.019, 0.996, 5 0.5 1.5 0.15 390 0.126 ± 0.005 0.022 ± 0.013 0.02, 0.996, 5 0.5 1.6 0.16 100 µl NR2, 100 µl NaOH, 0.708 mM tartrato, H2O nanopura tratada. 600 0.127 ± 0.005 0.023 ± 0.013 0.02, 0.996, 5 0.5 1.6 0.16 0 (0.111 ± 0.007) (0.062 ± 0.019) 0.03, 0.988, 5 0.8 2.7 0.27 210 (0.117 ±0.004) (0.089 ± 0.011) 0.017, 0.996, 5 0.4 1.5 0.15 390 (0.116 ± 0.004) (0.098 ± 0.011) 0.019, 0.996, 5 0.5 1.6 0.16 100 µl NR2, 100 µl NaOH, 0.708 mM tartrato, 100 µl PVA 600 (0.115 ± 0.005) (0.103 ± 0.013) 0.02, 0.995, 5 0.5 1.8 0.18
- 107. Capítulo 2. RESULTADOS OBTENIDOS Y DISCUSIÓN 83 Revisión del Método de Electrodo Selectivo Las curvas de calibrado de amonio, E(mV) frente a Log ( + 4NH C ), dieron una respuesta lineal como la que se muestra en la Figura 31-1. Las respuesta de las aminas utilizando este método fue polinómica, y diferente para cada amina (Figura 32 a). Además la respuesta de las aminas en presencia de amonio, fue diferente a la obtenida sin amonio (Figura 32 b). Por otra parte, la linealidad de la curva de calibrado de amonio se perdió en presencia de una concentración fija de amina ajustándose los datos a una función polinómica, como puede verse en la Figura 31-2. Se concluyó que el método del electrodo selectivo de amonio, presentaba error sistemático en presencia de aminas. Figura 32. Curvas de calibrado de: a) Aminas. b) Aminas en presencia de 1 mg/l de amonio. (♦ metilamina, etilamina, isopropilamina, ×× β-feniletilamina) Figura 31. Curvas de calibrado de: 1-Amonio. 2-Amonio en presencia de MA (1 mg/l). -140 -120 -100 -80 -60 -40 -20 0 20 40 60 -1.5 -0.5 0.5 1.5 2.5 LOG(NH4+) E(mV) 1 2 -60 -40 -20 0 20 40 60 80 100 0 5 10 15 20 C amina (mg/L) E(mV) a) -60 -50 -40 -30 -20 -10 0 10 0 5 10 15 20 C amina (mg/L) E(mV) En presencia de 1 mg/L NH4+ b)
- 108. REACCIÓN DEL TCPO 84 Análisis de muestras de agua Aplicación del Método de Nessler Mediante un test t (á = 0.05) se comprobó que el tratamiento de decloración y precipitación de los cationes de las muestras, no afectaba a los resultados, siendo la pendiente de la curva realizada con patrones tratados, similar a la pendiente de la curva de patrones sin tratamiento (ver en Tabla 30). Para evaluar la presencia o ausencia de errores sistemáticos, se aplicaron el MOSA y el Youden a tres muestras de agua. Al aplicar el MOSA, las pendientes fueron en todos los casos estadísticamente diferentes a la pendiente de la curva de calibrado de patrones. Para las tres muestras, las pendientes variaban en función del volumen de muestra estudiado y del tratamiento aplicado. Este comportamiento se refleja en la Figura 33. Figura 33. Pendientes de las curvas de calibrado al aplicar el Método de Nessler en las diferentes condiciones estudiadas. (♦) Agua de riego (S1) ( ) Agua residual (S2) ( ) Agua de fuente (S3). Condiciones: NR1 o NR2 (Reactivo de Nessler 1 o 2); T: Muestra Tratada; NT: Muestra no tratada; TNC: Muestra tratada sin declorar; V: Volumen de muestra (mL); Volumen total= 2.5 mL. El estudio del error sistemático constante mediante el método de Youden [56], dio una ordenada en el origen diferente de cero para la muestra de agua residual (S2) desde t = 210 s a t = 390 s, por lo que el valor del TYB (Blanco Total de Youden) se tuvo en cuenta al calcular la concentración de amonio en la muestra. La Figura 34, muestra este resultado. 0 0.1 0.2 0.3 0.4 0.5 0.6 NT,V=2.4 TNC,V=2.4 T,V=2.4 T,V=2.4 T,V=1.5 T,V=1.5 T,V=0.05 NT,V=0.05 T,V=0.1 T,V=0.1 T,V=0.2 T,V=2.2 Condiciones (b±sb) NR1 NR2 Valor de Referencia
- 109. Capítulo 2. RESULTADOS OBTENIDOS Y DISCUSIÓN 85 Figura 34. Gráfico de Youden a diferentes tiempos para la muestra de agua residual. De estos estudios, se concluyó que al utilizar el Método del Reactivo Nessler para calcular la concentración de amonio en muestras de agua reales, habría de aplicarse el Método de Adición Estándar (MOSA) teniendo en cuenta el valor del TYB cuando fuese necesario, o aplicar el GHPSAM [57], que permite determinar la concentración del analito en muestras desconocidas eliminando ambos errores sistemáticos a la vez. Las concentraciones de amonio encontradas en las tres muestras estudiadas utilizando este método aplicando el MOSA o el GHPSAM, se muestran en la Tabla 31. Estas concentraciones responden a las concentraciones habituales de amonio en estos tipos de muestras. Aplicación del Método de Electrodo Selectivo En la bibliografía [58] se recomendaba aplicar el método de adición estándar si la muestra presentaba cantidades muy bajas de amonio, por lo que se aplicó el MOSA a la muestra de agua de riego. La pendiente del MOSA (b ± sb) = (-56.3 ± 0.5), resultó estadísticamente igual a la pendiente de la curva de calibrado con patrones (b ± sb) = (-6.6 ± 1.2). El cálculo de la concentración de amonio en las muestras, se realizó por medida directa de la señal e interpolación en la curva de calibrado de patrones. Los resultados se muestran en la Tabla 31. -0.2 -0.1 0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9 0 0.05 0.1 0.15 0.2 0.25 Volumen de muestra (ml) Absorbancia t=0s t=390s t=300s t=210s t=90s
- 110. REACCIÓN DEL TCPO 86 Tabla 31. Concentración de amonio hallada en muestras reales utilizando los diferentes métodos estudiados. Muestra Método Por Tiempo (s) Conc (mg/l) (C ±± s) Medida directa - 0.0901 Medida directa - 0.0903 Electrodo Selectivo Medida directa - 0.0867 (0.089 ± 0.002) MOSA 210 0.1545 MOSA 300 0.1725 MOSA 390 0.1721 (0.166 ± 0.010) GHPSAM 210 0.1733 GHPSAM 300 0.2253 Nessler GHPSAM 390 0.1733 (0.19 ± 0.03) MOSA (λexc = 415nm) 300 0.1984 S1 OPA-NAC MOSA (λexc = 333nm) 120 0.1688 (0.18 ± 0.02) Medida directa - 15.6375 Medida directa - 16.9618 Electrodo Selectivo Medida directa - 17.6654 (16.8 ± 1.0) MOSA 210 17.3592 MOSA 300 18.1065 MOSA 390 18.5709 (18.0 ± 0.6) GHPSAM 210 16.75 GHPSAM 300 - Nessler GHPSAM 390 17.5 (17.1 ± 0.5) MOSA (λexc = 415nm) 300 14.2195 S2 OPA-NAC MOSA (λexc = 415nm) 300 13.9740 (14.10 ± 0.17) Medida directa - 0.4229 Medida directa - 0.4587 Electrodo Selectivo Medida directa - 0.4587 (0.45 ± 0.02) MOSA 210 0.3014 MOSA 300 0.3379Nessler MOSA 390 0.3297 (0.323 ± 0.019) MOSA (λexc = 415nm) 300 0.3842 S3 OPA-NAC MOSA (λexc = 333nm) 120 0.3523 (0.37 ± 0.02) Aplicación del Método de OPA-NAC La aplicación de Método de Youden dio ordenada en el origen estadísticamente igual a cero a λexc = 415 nm para las tres muestras, mientras que a λexc = 333 nm, la muestra de agua residual presentó error sistemático constante.
- 111. Capítulo 2. RESULTADOS OBTENIDOS Y DISCUSIÓN 87 Las curvas de calibrado obtenidas al aplicar el MOSA (Tabla 32), mostraron que a λexc = 415 nm, ninguna de las tres muestras presentaba error sistemático proporcional, por lo que la concentración podía deducirse a partir de la curva de calibrado de patrones. Por otra parte, a λexc = 333 nm las muestras S1 y S3 estaban libres de errores sistemáticos proporcionales, pero la muestra de agua residual presentó efecto matriz. Por tanto, para calcular la concentración de la muestra 2, habría que utilizar los datos a λexc = 415 nm. Las concentraciones de amonio halladas en las muestras, se reflejan en la Tabla 31. Tabla 32. Curvas de adición estándar para muestras reales utilizando el Método OPA-NAC. Muestra λλex (nm) a ±± sa b ±± sb (n, r 2 , sy/x) S1 415 103 ± 17 710 ± 20 (6, 0.9958, 30) S1 333 550 ± 50 1380 ± 70 (6, 0.99, 90) S2 415 466 ± 4 688 ± 5 (6, 0.9997, 7) S2 333 3999 ± 16 480 ± 20 (5, 0.9943, 30) S3 415 62 ±12 803 ± 17 (6, 0.9983, 20) S3 333 540 ± 40 1440 ± 60 (6, 0.9932, 80) Comparación de resultados Los resultados de la Tabla 31, se compararon mediante un test t de muestras relacionadas, obteniéndose siempre valores de tcalc < ttab siendo los valores de á superiores a 0.05 : 0.213, 0.190, 0.113 para las comparaciones OPA-NAC frente a Electrodo Selectivo, OPA-NAC frente a Nessler y Electrodo Selectivo frente a Nessler, respectivamente. Con estos valores, se concluyó que los resultados eran similares en los tres métodos estudiados. En la Tabla 33 se reflejan las principales propiedades de los tres métodos. Se observa que los límites de detección y cuantificación que se obtuvieron al utilizar el Método OPA-NAC fueron inferiores a los obtenidos con los métodos de Nessler o Electrodo selectivo. El intervalo dinámico de concentraciones para el Método OPA-NAC abarca concentraciones más bajas. Los estudios de precisión dieron valores de desviación estándar relativa (DER%) similares para los tres métodos: 15-3 %, 16-5% y 18-1% para el Nessler, electrodo selectivo y OPA-NAC respectivamente. El Método de Nessler presentó la desventaja de la aparición de un precipitado cuando se trabajó con muestras reales, y además se detectaron errores sistemáticos constantes y proporcionales, que requerían la aplicación del MOSA y el Youden, o bien la aplicación del GHPSAM, para el cálculo de la concentración de amonio en las muestras. Por su parte, el Método del Electrodo Selectivo, permitió determinar el amonio por medida directa de la muestra, aunque presentaba la desventaja de la interferencia de aminas con señal no aditiva. Por último, el Método OPA-NAC, no presentó efecto matriz,
- 112. REACCIÓN DEL TCPO 88 pudiéndose calcular la concentración de amonio a partir de la curva de calibrado de patrones. Este método resultó además selectivo a ëexc = 415 nm. Tabla 33. Propiedades analíticas de los tres métodos estudiados. MétodosPropiedades analíticas Nessler Electrodo Selectivo OPA/NAC Intervalo dinámico 0.85-5 mg/L 5 -100 mg/L 0.2 -1.4 mg/L Límite de Detección 0.6 mg/L 1.6 mg/L 0.07 mg/L Reproducibilidad ** ** ** Selectividad Método selectivo Interferencia de aminas Método selectivo Coste ** * ** Rapidez 4 muestras/hora 7 muestras/hora 7 muestras/hora En los tres métodos las señales analíticas variaron en función del tiempo. En el Método de Nessler, la señal se midió a los 10 minutos para asegurar una señal estable. En el método del electrodo selectivo, la señal se midió cuando el valor del potencial permanecía estable durante 1 minuto. En el método de OPA-NAC, el tiempo de reacción fueron 5 minutos. Teniendo en cuenta todas estas consideraciones, se concluyó que el método de OPA-NAC podía utilizarse para la determinación de amonio aportando rapidez, selectividad y sensibilidad. El límite de detección alcanzado está por debajo del requerido por el Official Journal of European Communities para establecer la calidad del agua de consumo humano. Conclusiones En este trabajo, se propuso el uso de la mezcla OPA-NAC como reactivo derivatizante para el amonio. El isoindol fluorescente formado se detectó selectivamente en presencia de aminas excitando a 415 nm. El procedimiento optimizado se comparó con el método de Nessler y el método de Electrodo Selectivo, que fueron revisados previamente a su utilización. La aplicación del método de Nessler a muestras reales presentó algunos problemas como la formación de precipitado y el efecto matriz. Para calcular la concentración de amonio, se requirió la aplicación del MOSA o del GHPSAM. Por su parte, la aplicación del método del Electrodo Selectivo a muestras reales presentó el problema de la interferencia de aminas con señal no aditiva. Sin embargo, al utilizar el método de OPA-NAC, la concentración de amonio se podía calcular a partir de la curva de calibrado de patrones, aporta mejores límites de detección y presenta una velocidad de muestreo similar a la del electrodo selectivo.
- 113. Capítulo 2. RESULTADOS OBTENIDOS Y DISCUSIÓN 89 2.1.2.2. DETERMINACIÓN QUIMIOLUMINISCENTE DE AMONIO EN AGUAS POR HPLC. Los métodos de referencia para la determinación de amonio, Nessler, el indofenol y electrodo selectivo, presentan problemas de selectividad [55,59-61]. En la bibliografía se han descrito algunos procedimientos para mejorarla. En estos trabajos, se ha utilizado una resina de intercambio aniónico ligeramente básica [62] o bien una columna de intercambio catiónico de alta capacidad [63], para separar el amonio de otros cationes inorgánicos y/o aminas alifáticas que pueden presentar las muestras de agua. Existen también métodos para medir amonio basados en las reacciones de derivatización con o-ftaldialdehido (OPA), como el reactivo OPA-sulfito [59] o más recientemente el OPA-tioglicolato [64] y el OPA-NAC (descrito en el capítulo precedente, Apéndice 4 [55]) por formación de derivados fluorescentes, que si bien presentan mejores prestaciones que los anteriores, pueden conducir a errores sistemáticos en algunas ocasiones, ya que la fluorescencia del fondo debida a compuestos orgánicos naturales no suele ser insignificante en aguas naturales [59]. También se han desarrollado procedimientos quimioluminiscentes para la determinación de amonio [65-66] debido a su elevada sensibilidad, rapidez, simplicidad y viabilidad. El reactivo utilizado normalmente es el luminol combinado con el hipoclorito, en sistemas de inyección en flujo, pero no ha sido descrita su aplicación a HPLC para mejorar su selectividad. Aunque la reacción entre los compuestos de nitrógeno y el cloruro de dansilo (DnsCl) es bastante conocida, no se ha aplicado a la determinación cuantitativa de iones amonio con sistema de detección fluorimétrico o con generación quimioluminiscente post-columna (con reactivos como Bis-(2,4,6-triclorofenil)oxalato, TCPO). Sin embargo, la reacción de dansilación se ha estudiado ampliamente para la determinación de analitos como aminas biogénicas en plantas [67], aminas de bajo peso molecular en muestras de agua como se ha descrito en capítulos precedentes [68,21,36], fenoles en aguas superficiales [69], residuos de pesticidas del tipo fenoxil N-metilcarbamato en zumos de frutas [70] y anfetaminas en muestras de orina [71,18]. Basándonos en la experiencia de nuestro grupo de investigación en la derivatización de aminas con Cloruro de Dansilo y TCPO [36,71,18], se desarrolla en este trabajo un método quimioluminiscente rápido, sensible y selectivo para el screening y cuantificación de amonio en muestras de agua de baja concentración de amonio (<0.5 mg/l), pero también aplicable a muestras de concentración más elevada. El método se basa en la derivatización directa de amonio en la muestra (sin ningún tratamiento previo) con el reactivo cloruro de dansilo, seguido de HPLC para separar la señal analítica del amonio de la respuesta de otros compuestos de nitrógeno. Después de la separación en una columna analítica C18, los derivados se mezclaron con el TCPO/H2O2 y se registró la señal quimioluminiscente.
- 114. REACCIÓN DEL TCPO 90 Se estudió la interferencia de los derivados de aminas en la señal del derivado de amonio, y también se propuso un procedimiento para el screening de aminas alifáticas y amonio en muestras de agua de diferente naturaleza. Además, el método se aplicó a varias muestras después de aplicar el tratamiento Kjeldahl, para obtener valores de Nitrógeno Kjeldahl Total. Más información acerca de las condiciones experimentales se puede encontrar en el Apéndice 5. Optimización de la dansilación precolumna del amonio La dansilación convencional de aminas se basa en la reacción directa con cloruro de dansilo en presencia de una concentración elevada de bicarbonato, y algunos autores recomiendan que la reacción transcurra durante toda la noche (12 horas a temperatura ambiente). En este trabajo, el amonio se derivatizó de acuerdo con las condiciones establecidas por Marcé y col. para poliaminas [67], con algunas modificaciones introducidas para hacer más rápido el procedimiento. Para establecer las condiciones óptimas, los derivados dansilados se inyectaron en el sistema cromatográfico, y la reacción quimioluminiscente post-columna se llevó a cabo siguiendo el procedimiento descrito por nuestro grupo de investigación para aminas alifáticas previamente desarrollado [36] (ver el montaje utilizado en la Figura 15, Pág. 39 del Capítulo 1- Introducción). El derivado dansilado de amonio eluyó a un tiempo de retención de 2.4 minutos (Figura 35). El cromatograma del blanco presentó un pico al mismo tiempo de retención que el pico del amonio. Este pico aparecía en mayor o menor medida en función de la cantidad de disolvente empleado (Acetonitrilo 1, Scharlau) en la disolución del reactivo. Se intentó separar este pico del pico del derivado de amonio, mediante la utilización de diferentes gradientes de elución y también utilizando una precolumna con diferentes tipos de relleno (CN, C18, SCX, o SAX) insertada mediante una válvula de conmutación, pero en ningún caso se obtuvo separación, lo que indicaba que el pico se debía a una contaminación de amonio en el disolvente empleado. Por ello, se ensayaron diferentes disolventes: acetona, acetonitrilo 2 (J.T.Baker), tetrahidrofurano y metanol. La mezcla del reactivo contenía 0.9 mL de disolvente (0.72 mL más 0.18 mL de disolución de reactivo DnsCl 5 mM), 0.2 mL de tampón carbonato 0.1 M de pH 9 y 1 mL de agua nanopura o patrón de amonio. Aunque con disolventes como tetrahidrofurano (THF) o metanol, no apareció pico de blanco, ninguno de ellos fue apropiado. El metanol disminuyó mucho la eficiencia de la reacción post-columna, y el THF impidió la reacción de dansilación al encontrarse el reactivo DnsCl y el NH4 + en dos fases diferentes. La acetona presentó el mismo comportamiento que el acetonitrilo 1, siendo apropiada para llevar a cabo ambas reacciones (pre- y post- columna), pero presentaba un pico de blanco significativo. Al
- 115. Capítulo 2. RESULTADOS OBTENIDOS Y DISCUSIÓN 91 utilizar Acetonitrilo 2 (J.T.Baker), el pico del blanco no fue significativo, siendo la eficiencia de la reacción del amonio muy buena (Figura 35). Con estos resultados, se seleccionó Acetonitrilo 2 (J.T.Baker) para los siguientes experimentos. Figura 35. Cromatogramas del blanco y patrón de amonio de 0.187 mg/L en las condiciones óptimas de derivatización. Se ensayaron diferentes volúmenes de disolvente para seleccionar el mínimo necesario para llevar a cabo la reacción de derivatización sin perder la respuesta del amonio. En la Figura 36 se muestra la influencia del volumen de disolvente en la señal del blanco. Se seleccionaron 0.4 mL de acetonitrilo como volumen óptimo por proporcionar la menor señal del blanco sin pérdidas en la respuesta del amonio. La concentración del reactivo DnsCl en la mezcla de reacción, se estudió en el intervalo 0.15-0.75 mM, manteniendo constante el volumen de acetonitrilo en 0.4 mL. Se escogió como concentración de reactivo óptima 0.3 mM, pues proporcionaba la menor señal del blanco (ver Figura 36) sin que hubiera pérdidas en la respuesta del amonio. 0 1 2 3 4 Tiempo (min) SeñalQuimioluminiscente NH4+ Blanco Patrón
- 116. REACCIÓN DEL TCPO 92 Figura 36. Altura de pico del Blanco frente a Volumen de Acetonitrilo (Concentración de DnsCl fija 0.45 mM) o frente a concentración de DnsCl (Volumen de acetonitrilo fijo 0.4 mL). De acuerdo con estos resultados, las condiciones óptimas seleccionadas fueron las que se muestran en la Tabla 34: 0.4 mL de cloruro de dansilo 1.5 mM disuelto en Acetonitrilo (J.T.Baker), 0.05 mL de tampón carbonato 0.2 M pH 9 y 1.55 mL de patrón de amonio/muestra. En esta Tabla, se comparan las condiciones establecidas por Marcé y col. [67] con las optimizadas en este trabajo. Como puede verse, se simplificó el procedimiento original para hacerlo más rápido. Tabla 34. Comparación del procedimiento de derivatización en disolución establecido por Marcé y col.[67] y el procedimiento de derivatización en disolución optimizado. Procedimiento de Marcé y colaboradores Procedimiento optimizado en este trabajo 200 ìL muestra/patrón + 40 ìL patrón interno + 200 ìL de carbonato sódico saturado + 400 ìL DnsCl 5 mg/ml en acetona 1.55 mL muestra/patrón + 50 ìL de tampón carbonato 0.2 M pH 9 + 400 ìL DnsCl 1.5 mM en acetonitrilo Incubar en la oscuridad toda la noche a Tª ambiente o calentar 10 min a 70 ºC Calentar 10 minutos a 70ºC Añadir 100 ìL de Prolina de 100 mg/ml e incubar en la oscuridad 30 minutos Inyectar 20 ìL en el sistema cromatográfico Extraer los derivados dansilados en 500 ìL de tolueno y mezclar 30 segundos Separar 400 ìL de la fase orgánica, secar y redisolver en 800 ìL de acetonitrilo Pasar a través de un filtro de tamaño de poro 0.45 ìm Inyectar 20 ìL en el sistema cromatográfico 0 10000 20000 30000 40000 50000 60000 70000 80000 Volumen de Acetonitrilo (mL) o Concentración de DnsCl (mM) AlturadePicodelBlanco(V) Volumen de acetonitrilo Variable Concentración de DnsCl Variable Condiciones óptimas0.18 mL 0.4 mL 0.65 mL 0.9 mL 0.15 mM 0.3 mM 0.75 mM 0.45 mM
- 117. Capítulo 2. RESULTADOS OBTENIDOS Y DISCUSIÓN 93 Características analíticas En la Tabla 35 se muestra la curva de calibrado obtenida en las condiciones óptimas de derivatización. Se obtuvo buena linealidad en el intervalo 0.027-0.75 mg/L de amonio, y el límite de detección establecido como la concentración requerida para generar una relación (S/N)=3 fue de 8 µg/L. El cromatograma correspondiente al límite de detección aparece en la Figura 37-2. El límite de detección alcanzado por el método está muy alejado del límite establecido por la legislación (0.5 mg/L de amonio para muestras de agua no digeridas, y 2 mg/L par muestras de agua tratadas mediante el método de Kjeldahl). En la Tabla 35, también se muestran las curvas de calibrado obtenidas utilizando una concentración de reactivo menor (0.15 mM) y utilizando la concentración óptima de reactivo en presencia de diferentes salinidades. Al utilizar concentración de DnsCl 0.15 mM, la sensibilidad fue el 13% de la obtenida en las condiciones de concentración de reactivo óptimas. Los valores de la pendiente obtenidos en diferentes salinidades fueron estadísticamente similares a los obtenidos en ausencia de NaCl a un nivel de probabilidad del 95%. Por tanto, el método no estaba influenciado por la salinidad. Tabla 35. Ecuaciones de las curvas de calibrado (Altura de Pico(Voltios) frente a Concentración de NH4 + (mg/L)) obtenidas en diferentes condiciones. Condición DnsCl NaCl Curva de Calibrado Y = (a ±± sa) + (b ±± sb)· C (n, r 2 , sy/x) LD (µg/L) Intervalo Lineal (mg/L) 1 0.15 mM Y = (12 ± 6) + (130 ± 10)· C (5, 0.9814,9) 60 0.28-1 2 0.3 mM Y = (2 ± 7) + (960 ± 30)· C (10, 0.9935, 16) Y* = (3 ± 90) + (12300 ± 400)· C (10, 0.9929, 205) 8 0.027-0.75 3 0.3 mM 3.5 g/l Y = (14± 5) + (930 ± 30)· C (3, 0.9998, 3) 8 0.027-0.75 4 0.3 mM 35 g/l Y =(19 ± 14) + (980 ± 70)· C (3, 0.9942, 16) 8 0.027-0.75 *Area de Pico(Voltios) frente a Concentración de NH4 + (mg/L). En la Tabla 36 se muestran los estudios de precisión y exactitud del método a diferentes niveles de concentración. La reproducibilidad y repetibilidad del método fueron buenas independientemente de la concentración ensayada, obteniéndose valores de porcentaje de desviación estándar relativa (%DER) entre 2.6-7.2 % y 4-8.6 % respectivamente. La exactitud del método se evaluó analizando varios patrones de concentración conocida, y como puede verse en la Tabla 36, resultó satisfactoria, proporcionando errores relativos (%Er) entre 5-9.8%. Incluso a niveles de concentración de amonio muy bajos (0.028 mg/L), la exactitud del método fue buena con un error relativo del 6%.
- 118. REACCIÓN DEL TCPO 94 Tabla 36. Estudios de precisión (%DER) y exactitud (%Er) proporcionados por el método a diferentes niveles de concentración de amonio (n, número de réplicas). Reproducibilidad Repetibilidad Exactitud NH4 + (mg/L) %DER (n) NH4 + (mg/L) %DER (n) NH4 + (mg/L) %Er (n) 0.11 6.2 (5) 0.028 5.8 (4) 0.15 7.2 (4) 0.165 8.6 (5) 0.055 5 (4) 0.2 4.0 (3) 0.165 5.9 (3) 0.3 2.6 (4) 0.5 8.4 (3) 0.193 9.8 (3) Para evaluar la selectividad del método y conociendo la reactividad del Cloruro de Dansilo con compuestos de nitrógeno, se estudió la posible interferencia de las aminas en la señal de amonio. En los sistemas acuosos, las aminas se suelen encontrar a niveles de ìg/L, y muy raras veces a niveles de mg/L. La Figura 37-3 muestra el cromatograma obtenido para una mezcla de amonio y aminas (MA, EA, DMA, N-PrA, BA, DEA, PeA y HA), y como ha sido demostrado en capítulos anteriores, no se observó solapamiento entre los picos. Figura 37. Cromatogramas correspondientes a: 1. Blanco. 2. Patrón de amonio de 8µg/L (LD). 3. Mezcla de varias aminas contaminadas de amonio: MA 0.1 mg/L, EA 0.2 mg/L, N- PrA 0.3 mg/L, DMA 0.3 mg/L, BA 0.3 mg/L, DEA 0.5 mg/L, PeA 0.3 mg/L y HA 0.3 mg/L. Se fortificó un patrón de amonio con 0.1 mg/L de MA, DMA, DEA y PeA, (que son las aminas que hasta el momento habíamos encontrado en muestras de agua) para estudiar la posibilidad de consumo del reactivo por parte de las aminas, y el porcentaje de 0 2 4 6 8 10 SeñalQuimioluminiscente Tiempo (min) NH4+ R MA EA N-PRA + DMA BA DEA PEA HA R 1 2 3 LD NH4+
- 119. Capítulo 2. RESULTADOS OBTENIDOS Y DISCUSIÓN 95 recuperación de la señal del amonio fue del 89.3%, por lo que se consideró que no afectaba su presencia a la determinación de amonio aún encontrándose a niveles superiores a los que estas se presentan en aguas. Análisis de muestras de agua Se analizaron 10 muestras de agua reales, y en varias de ellas se encontró amonio. A modo de ejemplo, en la Figura 38, se muestran los cromatogramas por triplicado obtenidos para la muestras de agua residual (dilución 1:130) (A) y de lago (dilución 1:8.25) (B). Ambas presentaban amonio en diferentes concentraciones. Figura 38. Cromatogramas de las muestras de agua (3 réplicas). A) Agua residual. B) Agua de lago. Para validar la exactitud del método se fortificaron las muestras con concentraciones de amonio conocidas. Al aplicar el MOSA, las muestras de agua natural(S1), de grifo (S2), de fuente (S3), de riego 2 (S5), de lago(S6) y residual (S10), presentaron pendientes similares a las de la curva de calibrado con patrones (ver Tabla 35) a un nivel de confianza del 95% como lo demostró el test t [72] dado en la Tabla 37, por lo que no había efecto matriz y la concentración de amonio en las muestras podía calcularse a partir de la curva de calibrado de patrones con resultados exactos. Para la muestra S4 (agua de riego 1) la pendiente de la curva de calibrado del MOSA fue similar a la de la curva de calibrado de patrones a un nivel de confianza del 98%. Para las muestras de agua marina, solo S7 dio pendientes similares a un nivel de confianza del 99%, mientras que S8 y S9 presentaron efecto matriz como puede verse en la Tabla 37. En presencia de efecto matriz, la concentración de amonio debe calcularse aplicando el 0 1 2 3 4 Tiempo (min) SeñalQuimioluminiscente NH4+ NH4+ A) B)
- 120. REACCIÓN DEL TCPO 96 MOSA. Las ecuaciones del MOSA (ordenada: a (sa), pendiente: b (sb), r2 , n, syx) para las muestras S8 y S9 fueron: 57(5), 490(30), 0.9976, 4, 8.8 y 55(5), 320(30), 0.9707, 5, 8.6, respectivamente. Las pendientes del MOSA fueron inferiores a la pendiente de la curva de calibrado de patrones de amonio (Tabla 35). Tabla 37. Test t de comparación de pendientes de las ecuaciones del MOSA para todas las muestras evaluadas con la pendiente de la curva de calibrado de patrones de amonio. Los valores tabulados corresponden a un nivel de confianza del 95 % excepto *(98 %) y **(99 %). Muestra Fcalc Ftab Conclusión 1 tcalc ttab Conclusión 2 S1-Natural 3.988 17.443 Varianzas Homogéneas 1.086 2.7767 Pendientes similares S2-Grifo 6.150 799.5 Varianzas Homogéneas 2.940 3.1824 Pendientes similares S3-Fuente 3.205 864.16 Varianzas Homogéneas 0.614 2.7764 Pendientes similares S4-Riego 1 1.606 864.16 Varianzas Homogéneas 3.540 2.7764 3.7469* Pendientes similares S5-Riego 2 92.003 17.443 Varianzas no Homogéneas 2.024 12.603 Pendientes similares S6-Lago 44.31 17.443 Varianzas no Homogéneas 1.264 12.496 Pendientes similares S7-Marina 1 2.23 799.5 Varianzas Homogéneas 3.894 3.1824 5.84** Pendientes similares S8-Marina 2 5.36 17.443 Varianzas Homogéneas 5.960 2.7764 4.6041** Efecto matriz S9-Marina 3 5.421 17.443 Varianzas Homogéneas 8.137 2.7764 4.6041** Efecto matriz S3- Kjeldahl 1.659 647.79 Varianzas Homogéneas 0.071 4.3027 Pendientes similares S4- Kjeldahl 2.378 16.044 Varianzas Homogéneas 0.057 2.5707 Pendientes similares S6- Kjeldahl 2.726 647.79 Varianzas Homogéneas 2.311 4.3027 Pendientes similares S9- Kjeldahl 3.409 864.16 Varianzas Homogéneas 0.619 2.7764 Pendientes similares S10- Kjeldahl 1.571 799.5 Varianzas Homogéneas 1.179 3.1824 Pendientes similares En la Tabla 38 se muestran las concentraciones encontradas en la muestra a las dos concentraciones de amonio adicionado, y los errores para estas muestras fueron inferiores en todos los casos al 10.7% excepto en la muestra S7, que presentó pendiente del MOSA similar a la de la curva de calibrado de patrones a un nivel de confianza del 99% (para esta muestra los errores relativos fueron algo mayores). Los cromatogramas típicos de la muestra de agua de fuente fortificada con 0, 0.15 y 0.3 mg/L de amonio se muestran el la Figura 39.
- 121. Capítulo 2. RESULTADOS OBTENIDOS Y DISCUSIÓN 97 Tabla 38. Concentración de amonio encontrada en muestras reales fortificadas con amonio a diferentes niveles de concentración antes y después del tratamiento Kjeldahl. Amonio libre Después del tratamiento Kjeldahl NH4 + adicionado 0.15 mg/L NH4 + adicionado 0.3 mg/L NH4 + adicionado 0.15 mg/L NH4 + adicionado 0.3 mg/L Muestra Concentración encontrada (%Er) Concentración encontrada (%Er) S1-Natural 0.155 (3.3%) 0.268 (10.7%) S2-Grifo 0.141 (6%) S3-Fuente 0.135 (9.3%) 0.299 (0.3%) 0.138 (8%) 0.299 (0.3%) S4-Riego 1 0.145 (3.3%) 0.268 (10.7%) 0.141 (6%) 0.278 (7.3%) S5- Riego 2 0.135 (10%) 0.271 (9.7%) S6-Lago 0.145 (3.3%) 0.283 (5.7%) 0.169 (12.7%) 0.327 (9%) S7-Marina 1 0.189 (26 %) 0.354 (18%) S9-Marina 3 0.147 (2%) 0.291 (3%) S10-Residual 0.149 (0.3%) 0.304 (1.3%) 0.148 (1.3%) 0.320 (6.7%) Figura 39. Cromatogramas de la muestra de agua de fuente fortificada con 0, 0.15 y 0.3 mg/L de amonio. En la Tabla 39 se describe la respuesta de screening que proporciona el método propuesto tomando como referencia la concentración límite establecida en la legislación. Se encontró amonio en las muestras S3, S6, S7, S8, S9 y S10, pero solo el agua residual (S10) y el agua marina 1 (S7), superaron el límite establecido por la legislación (0.5 mg/L). En la Tabla 39 se muestra además la concentración de amonio hallada en las 0 2 4 6 SeñalQuimioluminiscente Tiempo (min) S2 S2 + 0.15 mg/l NH4+ S2 + 0.3 mg/l NH4+ NH4+ NH4+ NH4+
- 122. REACCIÓN DEL TCPO 98 muestras utilizando la curva de calibrado de patrones de amonio (Tabla 35) y utilizando el MOSA, observándose resultados similares. El agua de fuente y el agua residual se analizaron también utilizando el método de OPA-NAC anteriormente descrito (Apéndice 4, [55]). Los resultados fueron 0.37 (s=0.02, n=3) y 32 (s=0.4, n=3), respectivamente. Ambos valores pueden considerarse estadísticamente similares (a un nivel de confianza del 95%) a los obtenidos por el método propuesto. El amonio cuantificado en el agua residual debería observarse con reservas, debido a la gran dilución llevada a cabo en la muestra tanto por el método quimioluminiscente como por el fluorescente. Un grupo representativo de estas muestras se trató para evaluar el Nitrógeno Kjeldahl Total. El test t presentado en la Tabla 37, indicó que la pendiente del MOSA era similar a la de la curva de patrones de amonio. Por tanto, ninguna de las muestras presentaba efecto matriz, pudiéndose obtener la concentración de amonio a partir de la curva de calibrado con patrones. Para validar la exactitud, las muestras tratadas mediante el procedimiento Kjeldahl, se fortificaron con concentraciones conocidas de amonio, y las concentraciones de amonio halladas se muestran en la Tabla 38. Como puede observarse, se obtuvieron errores relativos bajos, entre el 0.3 y el 12.7%. La concentración de amonio en las muestras después de aplicar el tratamiento Kjeldahl, fue superior en todos los casos a la concentración del amonio libre (Tabla 39). Las muestras de agua de fuente, residual y de lago, dieron una respuesta de screening positiva, siendo su concentración superior al límite establecido por la legislación (2 mg/L). Tabla 39. Respuesta de screening y Concentración de amonio deducida para las muestras estudiadas. Muestra Límite legislado (mg/l) Respuesta Screening Concentración NH4 + (C ±± s) (mg/l) (n=3) Curva patrones Concentración NH4 + (C ±± s) (mg/l) (n) MOSA S1-Natural 0.5 No - - S2-Grifo 0.5 No - - S3-Fuente 0.5 No (0.367 ± 0.012) 0.347 (n=5) S4-Riego 1 0.5 No - - S5-Riego 2 0.5 No - - S6-Lago 0.5 No (0.352 ± 0.014) 0.378 (n=5) S7-Marina 1 0.5 Si (3.8 ± 0.1) 3.108 (n=5) S8-Marina 2 0.5 No Efecto matriz 0.098 (n=5) S9-Marina 3 0.5 No Efecto matriz 0.147 (n=5) S10-Residual 0.5 Si (30 ± 4) 30.69 (n=4) S3 Kjeldahl 2 Si (2.28 ± 0.12) 2.34 (n=3) S4 Kjeldahl 2 No (0.56 ± 0.10) 0.595 (n=4) S6 Kjeldahl 2 Si (3.05 ± 0.13) 2.71 (n=3) S9 Kjeldahl 2 No (0.81 ± 0.08) 0.885 (n=5) S10 Kjeldahl 2 Si (169 ± 18) 152.6 (n=4)
- 123. Capítulo 2. RESULTADOS OBTENIDOS Y DISCUSIÓN 99 Conclusiones En este trabajo se estudió la detección quimioluminiscente del amonio con la intención de mejorar la selectividad y sensibilidad de su determinación. Se utilizó el procedimiento de dansilación en disolución. El derivado dansilado se separó mediante un sistema de HPLC y se mezcló post-columna con los reactivos TCPO/H2O2 generándose la quimioluminiscencia. Se consiguió la cuantificación de amonio a niveles bajos (0.027-0.75 mg/L) con buena exactitud (errores inferiores al 9.8%) y precisión (%DER entre 2-8.6%). El método resultó selectivo frente a la presencia de aminas en la muestra y puede aplicarse para el screening y cuantificación del amonio en muestras de agua, siendo el límite de detección de 8 ìg/L, nivel que está muy alejado del límite establecido por la legislación (0.5 mg/L para muestras de agua no digeridas, y 2 mg/L para muestras de agua tratadas mediante el procedimiento Kjeldahl). El amonio se pudo cuantificar a partir de la curva de calibrado de patrones para casi todos los tipos de muestras de agua analizados: embotellada (natural), de grifo, de lago, de fuente, de riego y residual. Sin embargo, dos de las tres muestras de agua marina ensayadas, requirieron la aplicación del método de adición estándar para la cuantificación del amonio debido a la presencia de efecto matriz, efecto que desapareció al aplicar el tratamiento Kjeldahl.
- 124. REACCIÓN DEL TCPO 100 2.2. MÉTODO DE SCREENING DE MUESTRAS PARA Cu(II). La necesidad de respuestas analíticas rápidas y fiables es incuestionable. Los métodos de screening de muestras [73-74] proporcionan una respuesta binaria Si/No, que indica si los analitos en cuestión están presentes en la muestra a un nivel de concentración por encima o por debajo de un nivel de corte establecido. Con esto, se consiguen algunas ventajas, como la reducción de los costes, rapidez, simplicidad y minimización de errores debido a las diferencias entre el muestreo y el análisis. Los métodos de screening tienden a dar información cualitativa más que cuantitativa, requieren muy poco o ningún tratamiento de la muestra, son rápidos y normalmente requieren confirmación mediante el uso del método convencional para las muestras cuyo resultado ha sido positivo. Ha de establecerse el límite de detección, el valor de corte alrededor del cual se establecerán las posibilidades de error del método, el umbral o valor establecido por el cliente o la legislación (marcará la respuesta Sí/No), y las incertidumbres de los valores anteriores.
- 125. Capítulo 2. RESULTADOS OBTENIDOS Y DISCUSIÓN 101 2.1.3.1. MÉTODO AUTOMÁTICO DE INYECCIÓN EN FLUJO PARA EL SCREENING DE MUESTRAS DE AGUA PARA Cu(II), BASADO EN LA REACCIÓN QUIMIOLUMINISCENTE DE COPROPORFIRINA I-Cu (II) / TCPO / H2O2. Igarashi y col. [75] propusieron un método para la determinación de Cu(II) basado en la reacción quimioluminiscente de la Coproporfirina I-Cu(II)/TCPO/H2O2 en estático. Basándonos en estos estudios, se propone en este capítulo un método automático para el screening de muestras de agua para Cu (II), basado en la reacción quimioluminiscente del peroxioxalato. La Coproporfirina I es el fluoróforo. En presencia de Cu(II), por efecto quenching, la emisión quimioluminiscente disminuye. Se utilizó un detector quimioluminiscente simple diseñado para inyección en flujo. Se analizaron varias muestras de agua y se validó el método mediante el análisis de un patrón de referencia certificado, verificándose su utilidad para cumplir con la legislación vigente sobre Cu (ver Tabla 3, Pág. 14 del Capítulo1-Introducción) El montaje de inyección en flujo utilizado es el que se muestra en la Figura 16 Pág. 40 del Capítulo 1-Introducción. Más información acerca de las condiciones experimentales se incluye en el Apéndice 6. Estudio de la señal del Detectorde Quimioluminiscencia FireFly Se evaluó la sensibilidad del detector utilizando el procedimiento en flujo descrito para la reacción del Cr(III)/Luminol/H2O2 [76]. Las curvas de calibrado Log-Log y los límites de detección obtenidos por nuestro grupo de investigación en publicaciones recientes [76-78] utilizando el espectrofluorímetro Hitachi F-4500, así como la curva obtenida con este detector, se muestra en la Tabla 40. El detector de Quimioluminiscencia FireFly presentó sensibilidad similar al Hitachi F-4500, con la ventaja de ser mucho más simple y presentar mucho menor coste. Tabla 40. Comparación de sensibilidad de los detectores en la reacción QL Cr(III)/luminol/H2O2. Referencia (loga ±± sloga) (b ±± sb) Límite de detección (µµg/l) Detector [18] (1.91 ± 0.03) (1.19 ± 0.03) 1.3 Hitachi F4500 [19] (1.97 ± 0.07) (1.25 ± 0.07) 1.2 Hitachi F4500 [20] (1.84 ± 0.03) (1.16 ± 0.03) 1.6 Hitachi F4500 Este trabajo (1.04 ± 0.06) (1.25 ± 0.06) 1.4 FireFly QL
- 126. REACCIÓN DEL TCPO 102 Optimización del sistema de flujo La medida del espectro de emisión del blanco dio un máximo a 622 nm (Figura 40 A). Una vez conocido el máximo de emisión, se registró la curva cinética de la reacción. En tan solo 10 segundos, se formaba el intermedio correspondiente y se desactivaba, como se muestra en la Figura 40 B. Figura 40. A) Espectro de emisión QL del blanco de Coproporfirina I. B) Curva cinética de la reacción QL Coproporfirina I/TCPO/H2O2. La optimización del sistema se llevó a cabo con la disolución blanco de Coproporfirina I, considerando óptimas las variables que maximizaban su señal, ya que el Cu(II) contribuye a su disminución. En la Tabla 41 se muestran las variables del sistema optimizadas, el intervalo de estudio y el valor óptimo seleccionado. Tabla 41. Optimización de las variables químicas y físicas del sistema de inyección en flujo. Variable Intervalo estudiado Óptimo seleccionado Concentración CTAC 2· 10 -5 – 9· 10 -2 M 1· 10 -2 M Concentración H2O2 0.1 – 1 M 0.7 M Concentración TCPO 1 – 10 mM 5 mM % MeCN:THF 0%:100% – 100%:0% 75%:25% pH 4.7-10.3 7.4 V flujo Bomba 2 10 - 48 rpm 40 rpm Volumen Bucle 50 – 500 µl 50 µl Distancia de la válvula de inyección (IV) al detector (QLD) LIV1-QLD= 12 - 24 cm LIV2-QLD=7.5 - 15 cm LIV1-QLD= 12 cm LIV2-QLD=7.5 cm Longitud de la guía de ondas 17 – 50 cm 17 cm Tiempo de reacción Coproporfirina I-Cu(II) 1 – 10 min 5 min 0 0.2 0.4 0.6 0.8 1 400 500 600 700 λλ (nm) SeñalQL A) Máximo 622 nm 0 20 40 60 80 100 0 10 20 30 40 50 60 70 80 90 Tiempo (s) SeñalQL B)
- 127. Capítulo 2. RESULTADOS OBTENIDOS Y DISCUSIÓN 103 En cuanto a las variables químicas estudiadas, destacar que la presencia de CTAC sensibilizó la reacción por ofrecer un entorno más adecuado. El H2O2 y el TCPO debían llegar al detector por separado para alcanzar la máxima sensibilidad. Sus concentraciones se variaron de acuerdo con los estudios recientes de esta reacción quimioluminiscente [79-80]. El TCPO se preparó en acetonitrilo:tetrahidrofurano 75:25 para evitar su degradación y obtener la máxima sensibilidad. El pH óptimo para la reacción se obtuvo con tampón bórax 0.1 M a pH 7.4. En cuanto a las variables físicas, del estudio de la velocidad de la bomba 2, se fijó una velocidad de flujo para el TCPO de 2.9 ml/min, y para la Coproporfirina I, de 5 ml/min. El volumen del bucle seleccionado fue de 50 ìL. Volúmenes mayores dieron problemas de reproducibilidad y presión. Dada la rapidez de la reacción, parámetros como la distancia desde las válvulas de inyección al detector o la longitud de la guía de ondas fueron estudiados. En ambos casos, los valores óptimos fueron las mínimas distancias posibles. La sensibilidad aumentó 10 veces al acortar la distancia de las válvulas de inyección al detector, además de evitarse problemas de dispersión del bolo de muestra. Por su parte, al disminuir la longitud de la guía de ondas, la sensibilidad aumentó 2.5 veces. Con todas estas variables optimizadas, la concentración de Coproporfirina I empleada para la reacción con Cu(II) fue 1· 10-6 M, concentraciones inferiores no dieron reacción. Se incorporó el Cu(II) al sistema y se estudió el tiempo de reacción Cu(II)- Coproporfirina I en el baño de agua. El efecto quenching aumentaba al aumentar en tiempo de reacción, seleccionándose como óptimo 5 minutos. Sensibilidad y selectividad del método Se obtuvieron las curvas de calibrado de Cu(II) en diferentes condiciones de velocidad de flujo de los portadores de Coproporfirina I y CTAC/H2O2 (0.7/0.5 o 2.2/0.7) y diferentes condiciones de tiempos de reacción en los baños, t1 y t2 (min). Los resultados se muestran en la Tabla 42. Tabla 42. Curvas de calibrado de Cu(II) y parámetros analíticos en diferentes condiciones (IL: Intervalo lineal; LD: Límite de detección). vbomba1 (ml/min) (porph. / CTAC-H2O2) t1 t2 a ±± sa b ±± sb IL (µµg/l) LD (µµg/l) n, r2 , sy/x /b 0.7/0.5 5 2 4300 ± 40 -18.3 ± 0.7 0-125 17 12, 0.984, 5.38 0.7/0.5 5 2 4370 ± 110 -18.9 ± 1.3 0-125 17 8, 0.972, 8.96 0.7/0.5 5 5 6770 ± 30 -30.7 ± 0.4 0-125 10 5, 0.999, 1.32 2.2/0.7 4 0 3840 ± 20 -19.0 ± 1.0 0-32 17 4, 0.994, 1.22 2.2/0.7 5 0 4255 ± 18 -36.6 ± 1.0 0-32 8.7 4, 0.999, 0.58 2.2/0.7 5 0 5010 ± 50 -30.7 ± 0.8 0-100 10 10, 0.994, 2.83
- 128. REACCIÓN DEL TCPO 104 Las dos primeras curvas obtenidas en las mismas condiciones (velocidades de flujo 0.7/0.5 y t1/t2 5/2 minutos) mostraron la reproducibilidad del sistema, siendo el porcentaje de recuperación de la primera respecto a la segunda del 97%. Al utilizar velocidades de flujo 0.7/0.5 y t1=5 minutos, se observó un aumento de sensibilidad de 1.65 veces al aumentar t2. Al aumentar las velocidades de flujo (2.2/0.7), no se requirió un tiempo de parada en el segundo baño, obteniéndose una pendiente similar a los casos anteriores. Cuando el tiempo de reacción se incrementó en un minuto más, la sensibilidad fue casi el doble, obteniéndose el mejor límite de detección de 8.7 ìg/L, nivel que f ue 5.8 veces inferior al límite establecido por la legislación para Cu(II) en aguas (50 ìg/L). Los límites de detección se calcularon como 3· sblanco/b, siendo sblanco = 105.96 (n=9). La curva de calibrado en estas últimas condiciones de máxima sensibilidad, se obtuvo en el intervalo 1-100 ìg/L, que fue el intervalo lineal, pues a concentraciones superiores de Cu(II), la señal permaneció constante. Los registros de inyección en flujo obtenidos se muestran en la Figura 41. Figura 41. Registros de inyección en flujo de los patrones de la curva de calibrado de Cu(II). Los estudios de repetibilidad y reproducibilidad para un patrón de 15.9 y 17.3 ìg/l de Cu(II) respectivamente, dieron porcentajes de desviación estándar relativa (%DER) del 16% y 12%. Los porcentajes de recuperación fueron del 93% y 109% respectivamente. Estos valores están en concordancia con la precisión y exactitud requerida en análisis de trazas. 0 4 8 Tiempo (min) 0 2000 4000 6000 SeñalQuimioluminiscente 0 µg/l 31.8 µg/l 47.7 µg/l 79.4 µg/l 95.3 µg/l
- 129. Capítulo 2. RESULTADOS OBTENIDOS Y DISCUSIÓN 105 Para validar la exactitud del método, se utilizó además un Material de Referencia Certificado, SRM © 1640, de agua dulce (ver composición Pág. 121). El método dio una concentración de Cu(II) en el material de referencia de (96 ± 14) ìg/L (Valor de Referencia del SRM 1640: 85.2 ìg/L). El porcentaje de recuperación fue (113 ± 17) % (n=3), quedando demostrada la exactitud del método. Para comprobar la selectividad del método, se estudió el límite de tolerancia (concentración a la cual la señal se modificaba más de un 10%) de los siguientes iones metálicos: Na+ , Mg2+ , K+ , Ca2+ , Ag+ , Mn2+ , Ni2+ , Zn2+ , Al3+ , Sn2+ , Pb2+ , Hg2+ , Fe2+ , Fe3+ , Cd2+ , Cr3+ y Co2+ . Los iones Na+ , Mg2+ , K+ , Ca2+ , Ag+ , Mn2+ , Ni2+ y Zn2+ solo interfirieron a concentraciones muy elevadas, entre 5 y 500 mg/l, que no son habituales en muestras de agua. Al3+ , Sn2+ , Hg2+ , Cd2+ , Cr3+ y Co2+ tuvieron límites de tolerancia más bajos, pero en ningún caso superaron los límites establecidos por la legislación. Solamente Pb2+ , Fe2+ y Fe3+ dieron un límite de tolerancia por debajo de su límite establecido por la legislación, pero estaba por encima de su concentración habitual en aguas, por lo que el método fue suficientemente selectivo para la determinación de Cu(II). En la Tabla 43 se muestran los límites de tolerancia obtenidos, así como los límites establecidos por la legislación y su concentración habitual en aguas, para los iones más tóxicos estudiados. Tabla 43. Estudio de la interferencia de iones metálicos. Interferente Límite Legislado (µµg/L) Conc. en aguas naturales (µµg/L) Limite de Tolerancia (µµg/L) Al(III) 10 3000 Fe(III) 2000 500 2000 Fe(III) 2000 500 2000 Sn(II) 300 Pb(II) 50 1 30 Hg(II) 1 0.07 300 Cd(II) 5 0.03 30 Co(II) 0.05 30 Cr(III) 50 1 3000 Fiabilidad del método para el screening de muestras de agua para Cu(II) A concentraciones cercanas al límite de detección, se pueden producir un determinado porcentaje de falsos positivos y falsos negativos. Para tener un valor de referencia, se estableció un valor de corte a concentración 2LD. Se consideró un falso positivo cuando una muestra de concentración inferior al nivel de corte, daba una respuesta positiva. Se consideró un falso negativo cuando una muestra de concentración igual o superior al nivel de corte, daba una respuesta negativa.
- 130. REACCIÓN DEL TCPO 106 En la Figura 42, pueden verse los porcentajes de falsos positivos y negativos correspondientes a la medida de patrones de concentraciones cercanas al límite de detección: 0.5LD, 1LD, 1.5LD, 2LD, 3LD y 3.5LD ìg/L. Como puede observarse, a partir de 3.5LD, el porcentaje de falsos desaparece. Este valor está lejos del valor legislado (50 ìg/L), por lo que no se cometerán errores al utilizar el método para el screening de muestras. -50 -40 -30 -20 -10 0 10 20 30 40 50 4,3 8,7 13 17,3 26 30,3 %FP%FN µg/l Límite Detección Nivel de Corte 0.5DL 1DL 1.5DL 2DL 3DL 3.5DL Figura 42. Porcentajes de falsos positivos (%FP) y falsos negativos (%FN) en patrones de Cu(II) a concentraciones cercanas al límite de detección(DL). Aplicación a muestras de agua El método se aplicó al screening y cuantificación de Cu(II) en muestras de agua reales. De las seis muestras analizadas de forma directa, solamente la muestra de agua de fuente dio una respuesta positiva (Tabla 44). Tabla 44. Resultados obtenidos en el screening y determinación de Cu(II) en muestras de agua reales. Muestra Respuesta de screening Conc. en la muestra (C ±± s) (µµg/l) Cadicionada (µµg/l) %Rec ±± s S1, muestra sintética No - 31.77 92 ± 16 (n=6) S2, agua natural No - 31.77 102 ± 19 (n=3) S3, agua natural No - 31.77 100 ± 13 (n=4) S4, agua natural No - 31.77 117 ± 7 (n=4) S5, agua de grifo No - 50 83 ± 11 (n=3) S6, agua de fuente Si (61 ± 5) (n=8) 50 119 ± 12 (n=3)
- 131. Capítulo 2. RESULTADOS OBTENIDOS Y DISCUSIÓN 107 Los porcentajes de recuperación obtenidos al fortificar las muestras con Cu(II), fueron cercanos al 100%, como puede observarse en la Tabla 44. Estos resultados validaron la exactitud del método, ya confirmada con el estudio del patrón de referencia certificado. Conclusiones En este apartado de la Tesis, se propuso un sistema automático para la determinación de Cu(II) y el screening de muestras de agua. Se optimizaron las condiciones químicas y físicas de la reacción: CTAC 1· 10-2 M, H2O2 0.7 M, TCPO 5 mM, Acetonitrilo:THF 75:25, tampón bórax pH 7.4, velocidad de las bombas rápida puesto que la reacción se da en menos de 10 segundos, y distancias desde las válvulas de inyección al detector, las mínimas posibles para evitar dispersiones. El intervalo lineal de concentraciones fue de 10 a 100 ìg/L, siendo el límite de detección 8.7 ìg/L (muy por debajo del límite establecido por la legislación, 50 ìg/L). La precisión del método fue adecuada, siendo los valores de %DER del 12% al 16%. Los estudios de selectividad dieron límites de tolerancia superiores a las concentraciones legisladas en la mayoría de iones estudiados. En los casos en que fueron inferiores, estaban a niveles no habituales en muestras de aguas. La exactitud del método se evaluó mediante la fortificación de las muestras de agua analizadas, cuyos porcentajes de recuperación fueron cercanos al 100%. Además, el método se validó mediante un patrón de referencia certificado proporcionando también resultados bastante exactos. Además, se estudió la fiabilidad del método para el screening de muestras de agua y se vio que solo aparecían falsos positivos y negativos a niveles cercanos al límite de detección. Por tanto el método permitía filtrar las muestras cuyo contenido superaba el límite establecido por la legislación (50ìg/L) de un conjunto de muestras de estudio, con fiabilidad adecuada.
- 133. Capítulo 2. RESULTADOS OBTENIDOS Y DISCUSIÓN 109 2.2. REACCIÓN DEL LUMINOL 1.1. MEJORA DE LA EXACTITUD DE LA DETERMINACIÓN DE Cr(III), Cr(VI) Y Co(II). Validar un método de análisis consiste en verificar y documentar su validez, es decir, su adecuación a los requisitos establecidos por el usuario. Estos requisitos se denominan parámetros de calidad o características significativas de método. Pueden distinguirse tres tipos de parámetros de calidad: § Estadísticos: Precisión, exactitud, selectividad, límite de detección, intervalo dinámico y robustez. Este tipo de parámetros son los que son estudiados más a fondo, y se denominan parámetros analíticos o figuras de mérito. De entre ellos, la exactitud es el parámetro más difícil de garantizar. § Operativos: Rapidez, equipamiento necesario, instrumentación requerida, necesidades energéticas, complejidad en la preparación de la muestra, etc.. § Económicos: Inversión inicial, costes de mantenimiento, personal requerido, etc.. La validación de la exactitud de un método puede llevarse a cabo por diferentes vías: § Estudio de errores sistemáticos. § Comparación del resultado del método que se está ensayando con el(los) resultado(s) obtenido(s) utilizando otro(s) método(s) basado(s) en principios físico-químicos distintos. § Aplicación del método a un material de referencia certificado cuya composición en uno o varios analitos ha sido certificada por un organismo de prestigio. § Participación en un ejercicio de comparación interlaboratorio. La reacción quimioluminiscente del luminol con peróxido de hidrógeno, se ha utilizado para la determinación de iones metálicos como Cr(III) y Co(II), que actúan como catalizadores. En esta Tesis se estudian tres aspectos que inciden en la exactitud de la determinación de Cr(III), Cr(VI) y Co(II): § Elaboración de una guía para la eliminación de errores sistemáticos en la especiación de Cr(III) y Cr(VI). § Estudio de la influencia de los protocolos de acondicionamiento de muestras de agua en la detección quimioluminiscente de elementos traza. § Propuesta de un modelo de calibración multivariada aplicada a la determinación quimioluminiscente simultánea de cobalto y/o cromo.
- 134. REACCIÓN DEL LUMINOL 110 2.2.1.1. ELABORACIÓN DE UNA GUÍA PARA LA ELIMINACIÓN DE ERRORES SISTEMÁTICOS EN LA ESPECIACIÓN DE Cr(III) Y Cr(VI) EMPLEANDO LA REACCIÓN DEL LUMINOL CON H2O2. APLICACIÓN A MUESTRAS DE AGUA. La determinación de niveles traza en muestras de agua medioambientales requiere técnicas analíticas de elevada sensibilidad y selectividad. Para la determinación de cromo en muestras de agua, se han utilizado diferentes métodos, como la espectrofotometría, fluorimetría, fluorescencia de rayos X, espectrometría de absorción atómica, espectrometría de emisión atómica, cromatografía, métodos electroquímicos y análisis quimioluminiscente. La técnica quimioluminiscente proporciona métodos para el análisis de trazas que son atractivos debido a su elevada sensibilidad y bajo coste. En la bibliografía se han descrito diferentes procedimientos para la determinación de Cr(III) y Cr(VI) (previa reducción a Cr(III) con H2O2 en HCl) en muestras de agua basados en la reacción del luminol con peróxido de hidrógeno, tal como se muestra en la Tabla 45. En todos los casos, la señal de emisión se registra a 425 nm y se utiliza EDTA como agente complejante o separación cromatográfica para evitar interferencias de otros metales. En la Tabla 45, se refleja como los límites de detección varían en función del procedimiento utilizado. En este apartado se profundiza en la especiación de Cromo centrándose en la exactitud, para lo que se ha estudiado: § El paso de calibración, utilizando diferentes modelos: potencial o Log-Log e intervalo lineal, utilizando el sistema luminol/H2O2. § El efecto de algunos interferentes y la interferencia de la matriz en estas determinaciones en presencia de EDTA. § Una discusión acerca de la utilidad del método de adición estándar (MOSA) en función del modelo de calibración elegido. § Evaluación de la exactitud y precisión del procedimiento para la cuantificación de Cr(III) y Cr(VI). § Desarrollo de un protocolo a seguir para evitar errores sistemáticos en la especiación de cromo.
- 135. Capítulo 2. RESULTADOS OBTENIDOS Y DISCUSIÓN 111 Tabla 45. Estudios recientes basados en la reacción del luminol-H2O2 para cromo. Ref. Método Límite Detección (µµg/L) [81] Determinación de Cr(III) y Co. Separación cromatográfica. 15 [82] Determinación Cr(III) Inyección en Flujo. EDTA como agente complejante. 0.01 [83] Determinación de Cr(VI) y Cr(III). SO2 como reductor. Separación cromatográfica. Cr(III): 50 Cr(VI): 100 [84] Determinación de Cr(VI) y Cr(III). Inyección en flujo. H2O2 en HCl como agente reductor. EDTA como agente complejante. 0.01 [85] Determinación de Cr(III) y Cr(VI) por cromatografía iónica. Sulfito sódico como agente reductor. Cr(III):120 Cr(VI):90 [86] Determinación de Cr(III). Inyección en flujo. 500 [87] Determinación Cr(VI) y Cr(III) con separación cromatográfica y una segunda columna cromatográfica para la reducción de Cr(VI) a Cr(III). 2 [88] Determinación de Cr(III). Inyección en flujo. 5.2 El montaje de inyección en flujo utilizado es el que se muestra en la Figura 17 Pág. 40 del Capítulo 1-Introducción. Más información acerca de las condiciones experimentales se refleja en el Apéndice 7 [77]. Modelos de Calibración para Cr(III), Cr(VI) y Cr Total Las curvas de calibrado Señal-Concentración, siguen una ecuación potencial del tipo: S = a· Cb , que al tomar logaritmos se transforma en una recta del tipo: LogS = Loga + b· LogC. También se han obtenido las ecuaciones de las rectas de calibrado considerando únicamente el intervalo donde el comportamiento es lineal. Se ensayaron reactivos de diferentes calidades: HCl(p.a.) / NaHCO3(r.a.) / Na2CO3· 10H2O(p.a.), o bien HCl(trace pure) / Na2CO3(p.a.), y se observó que la señal del blanco desaparecía al utilizar los reactivos de mayor pureza: HCl(trace pure) / Na2CO3(p.a.). En la Tabla 46, se muestran las curvas de calibrado Log-Log e intervalo lineal para el Cr(III) y para el Cr(VI) utilizando reactivos para análisis de trazas, y en el caso del Cr(VI), a dos niveles de HCl diferentes (1· 10-3 M y 5· 10-3 M). Al observar los intervalos lineales, se dedujo que a mayor concentración de HCl, la sensibilidad era mayor, lo que significa que la concentración de HCl utilizada en la reducción de Cr(VI) a Cr(III) es un parámetro a controlar.
- 136. REACCIÓN DEL LUMINOL 112 Tabla 46. Curvas de calibrado para Cr(III) y Cr(VI). Curva de calibrado Cr HCl (M) Log-Log: a ±± sa; b ±± sb (n, sy/x, r 2 ) Intervalo lineal: a ± sa; b ± sb (n, sy/x, r 2 ) Cr(III)* - 1.06±0.07; 1.70±0.07 (10, 0.04, 0.9861) -330±70; 97±6 (10, 80, 0.9689) Cr(VI) 1· 10 -3 1.576±0.012; 1.403±0.013 (40, 0.02, 0.9967) -189±8; 115.9±1.1 (30, 17, 0.9976) Cr(VI) 5· 10 -3 1.927±0.008; 1.314±0.009 (40, 0.014, 0.9984) -252±17; 197±2 (30, 40, 0.996) En la Figura 43 se muestran los picos de inyección en flujo obtenidos para el calibrado de Cr(VI) con los reactivos de mayor pureza (HCl(trace pure) / Na2CO3(p.a.)), y concentración de HCl 5· 10-3 M para el tratamiento de reducción de Cr(VI) a Cr(III). Figura 43. Registros de señal para la curva de calibrado de Cr(VI) con HCl 5· 10 -3 M. Se llevó a cabo el estudio de mezclas de Cr(III) y Cr(VI), para examinar la posible determinación de Cr Total. En la Figura 44, puede observarse el solapamiento entre las curvas potenciales de Cr(VI) y Mezcla (Cr(VI)+Cr(III)) a dos niveles de concentración de HCl utilizado en el tratamiento de reducción (HCl 1· 10-3 M y HCl 5· 10- 3 M). Las curvas de calibrado potenciales de las mezclas dieron pendientes similares a las de las curvas de calibrado potenciales de Cr(VI), cuando se expresa la concentración de Cromo como Cromo Total (más información puede verse en el Apéndice 7, [77]). De estos resultados, se deduce que la conversión de Cr(VI) a Cr(III) después del tratamiento de reducción, es del 100 %. 0 1000 2000 3000 4000 Señalquimioluminiscente 0 g/L 3 g/L 6 g/L 10 g/L 15 g/L µ µ µ µ µ
- 137. Capítulo 2. RESULTADOS OBTENIDOS Y DISCUSIÓN 113 Figura 44. Curvas de calibrado potenciales para Cr(VI) (♦HCl 1· 10 -3 M, HCl 5· 10 -3 M) y para Mezclas (Cr(III)+Cr(VI))( HCl 1· 10 -3 M, ×× HCl 5· 10 -3 M). Por otra parte, al utilizar curvas de calibrado en el intervalo lineal, las pendientes fueron similares si se representaban frente a Cr(VI) adicionado o frente a Cr Total, lo que indicaba que en el intervalo lineal, Cr(III) y Cr(VI) se comportaban igual después del tratamiento de reducción. En la Tabla 47, pueden observarse estos resultados a los dos niveles de HCl utilizados para el tratamiento de reducción. Tabla 47. Curvas de calibrado en el intervalo lineal para las mezclas de Cr(III)+Cr(VI). Cr HCl (M) (a ±± sa) (b ±± sb) n r 2 Cr Total 1· 10 -3 (-270 ± 20) (129 ± 3) 20 0.9906 Cr (VI) adicionado 1· 10 -3 (120 ± 20) (129 ± 3) 20 0.9906 Cr Total 5· 10 -3 (-300 ± 50) (193 ± 4) 20 0.9932 Cr(VI) adicionado 5· 10 -3 (270 ± 20) (193 ± 4) 20 0.9932 Los calibrados obtenidos para mezclas (Figura 44, Tabla 47) fueron similares a los obtenidos para Cr(VI) (Tabla 46). Con esto, se concluyó que el método se puede aplicar para la especiación de Cromo en muestras de agua, determinando la concentración de Cr(III) previamente al tratamiento de reducción utilizando la correspondiente curva de calibrado, y después determinando el Cr Total utilizando la curva de calibrado preparada en las condic iones de reducción. El Cr(VI) se determinará por diferencia. Para evitar el uso de dos curvas de calibrado, se puede optar por acidificar las muestras con HCl y medir la CL del Cr(III) en estas condiciones. y = 37.695x 1.4028 R 2 = 0.9967 y = 84.534x 1.3144 R 2 = 0.9984 y = 46.845x 1.3307 R 2 = 0.9945 y = 90.704x 1.2507 R 2 = 0.9971 0 500 1000 1500 2000 2500 3000 3500 4000 0 5 10 15 20 Cr(VI) + Cr(III) (µµ g/l) SenyalCL
- 138. REACCIÓN DEL LUMINOL 114 En la Tabla 48 se muestran algunos parámetros analíticos correspondientes a las determinaciones de Cr(III), Cr(VI) y Cr Total. La precisión y exactitud fueron buenas, tal como se muestra en la Tabla 48. Tabla 48. Parámetros analíticos para las determinaciones de Cr(III), Cr(VI) y Cr Total. Precisión (%cv)** Exactitud (%Er)*** Cr, HCl (M) Cal* L.D. L.C. %cv5 %cv10 %cv15 % Er5 % Er10 % Er15 Cr(III) P L-L LI 1.9 1.9 2 4 4 4 12 2 12 9 1.4 9 10 1.4 10 5 5 5 4 4 5 5 5 6 %cv5.1 %cv10.3 %cv15.4 % Er5.1 % Er10.3 % Er15.4 Cr(VI), HCl1· 10-3 P L-L LI 1.1 1.1 1.9 3 3 4 4 0.6 4 3 0.4 3 5 0.7 5 1.3 0.2 5 3 0.5 5 2 0.3 1.5 Cr(VI), HCl5· 10-3 P L-L LI 1.8 1.8 1.8 4 4 4 3 0.5 4 1.3 0.17 3 2 0.3 5 2 0.4 5 7 0.9 3 5 0.6 0.9 %cv30.1 %cv35.3 %cv50.7 % Er30.1 % Er35.3 % Er50.7 Cr Total, HCl1· 10-3 P L-L LI 2 2 3 5 5 5 6 0.9 6 4 0.5 4 2 0.3 - 0.3 0.03 0.003 1.2 0.16 0.002 4 0.6 - Cr Total, HCl5· 10-3 P L-L LI 1.3 1.3 2 3 3 4 5 0.7 5 3 0.3 3 1.8 0.2 - 0.4 0.04 0.0014 0.11 0.017 0.0013 1.2 0.15 - * P: Curva de calibrado potencial. L-L: Curva de calibrado doble logarítmica. LI: Intervalo lineal. ** %cv: Coeficiente de variaciónconcentración adicionada (µg/l).. ***Er: Error relativoconcentración adicionada (µg/l). Estudio de interferencias Se demostró que el Cr(VI) no interfería en la determinación directa de Cr(III) en concentraciones inferiores a 10 mg/l. El Co(II) interfirió en la determinación directa de Cr(III) a niveles superiores a 50 µg/l, aumentando su interferencia después de aplicar el tratamiento de reducción debido a la presencia del HCl. Las curvas de calibrado de Cr(III)/Cr(VI) en presencia de Co(II): Directa (Cr(III)): y = (82 ±± 6)· C + (110 ±± 50) y después del tratamiento con HCl 5· 10-3 M (Cr(VI)): y = (175 ±± 9)· C + (2280 ±± 90), dieron pendientes similares a las obtenidas sin Co(II) (ver Tabla 46), lo que demostró la aditividad entre las señales de Cr y Co. En cuanto a los otros cationes y aniones estudiados: Cu(II), Mn(II), Ni(II), Mg(II), Ca(II), Fe(III), Cl- , Br- y SO4 2- , no produjeron interferencias.
- 139. Capítulo 2. RESULTADOS OBTENIDOS Y DISCUSIÓN 115 Aplicación a muestras reales S Método de adición estándar (MOSA) En la bibliografía no existen referencias acerca del uso de curvas potenciales para la evaluación del efecto matriz. Partiendo de datos simulados, se predijo cómo se modificarían los coeficientes a y b del modelo de curva potencial y=a· Cb . Se plantearon dos suposiciones: - Si no hubiese Cromo en la muestra, pero sí efecto matriz, un %Conversión (P) entre el 80-120 %, provocaría cambios en el coeficiente a, pero no en b, de forma que: y = a· (P· C)b = (a· Pb )· Cb = a’· Cb . Este efecto puede observarse en la Figura 45. Figura 45. Influencia del efecto matriz en las curvas potenciales. ♦Conversión 100%. Conversión 80 %. Conversión 120%. - Si la muestra contuviese Cromo, no podríamos utilizar las curvas potenciales porque no conoceríamos la concentración de Cromo Total, con lo que este modelo no sirve para evaluar el efecto matriz. Se estudió el comportamiento de un polinomio, y = a· X2 + b· X + c, al adicionar diferentes concentraciones de analito y en ausencia de efecto matriz. El coeficiente a se conserva, tal como puede verse en la Tabla 49, variando los coeficientes b y c. Los datos experimentales, no se adaptaron a este tipo de comportamiento, dando también una variación en el coeficiente a, tal como se observa en la Tabla 49. y = 24.88x1.2631 y = 32.985x1.2631 y = 41.526x1.2631 0 1000 2000 3000 4000 5000 6000 7000 8000 -15 5 25 45 65 Cromo Total SeñalCL
- 140. REACCIÓN DEL LUMINOL 116 Tabla 49. Curvas de calibrado simuladas y reales utilizando modelos de calibración polinómicos. Conc. Cr (µµg/l) Datos simulados Datos experimentales 0 y = 0.266· X2 + 105.7· X + 2402.2 y = 0.4688· X2 + 71.158· X - 134.16 5 y = 0.266· X2 + 108.36· X + 2937.4 y = 0.4076· X2 + 79.802· X + 182.53 10 y = 0.266· X2 + 111.02· X + 3485.8 y = 0.3646· X2 + 86.627· X + 557.03 15 y = 0.266· X2 + 113.68· X + 4047.5 y = 0.3056· X2 + 97.317· X + 1423 20 y = 0.266· X2 + 116.34· X + 4622.6 y = 0.266· X2 + 105.7· X + 2402.4 En el caso de que hubiese efecto matriz, en los datos polinómicos simulados, ninguno de los coeficientes del modelo se conservarían, con lo que se descartó para su evaluación. Con estos resultados, se concluyó que no se podían utilizar este tipo de curvas para la evaluación del efecto matriz, siendo únicamente posible diagnosticarlo si se utilizaban los intervalos lineales de concentración por comparación de las pendientes de calibración obtenidas. S Muestra de HCl El estudio de las rectas de adición estándar en una muestra de HCl que estaba contaminada con todos los interferentes estudiados, dio porcentajes de recuperación en pendiente respecto a la curva de calibrado de Cr(VI), entre el 94-114% cuando se trabajó en condiciones de reducción con HCl 5· 10-3 M y en el intervalo lineal de concentraciones. Al utilizar el volumen mayor de muestra (AE6) (ver Apéndice 7, [77]), el porcentaje de recuperación fue del 136%, por estar ya fuera del intervalo lineal (Tabla 50). El valor de la pendiente de la adición estándar sirve para garantizar que la muestra está dentro del intervalo lineal de Cromo establecido por el método. Tabla 50. Pendientes y porcentajes de recuperación para la muestra de HCl. Adición Estándar b ±± sb %Recuperación AE1 185 ± 5 93.9% AE2 200 ± 7 101.5% AE3 216 ± 3 109.6% AE4 225 ± 12 114.2% AE5 193 ± 4 98% AE6 268 ± 12 136%
- 141. Capítulo 2. RESULTADOS OBTENIDOS Y DISCUSIÓN 117 S Muestras de agua Se analizaron tres muestras de agua y un patrón de referencia certificado. Las adiciones estándar realizadas a dos volúmenes de una muestra de agua de grifo en condiciones de reducción con HCl 5· 10-3 M, dieron pendientes similares a las curvas de calibrado de patrones: (165 ± 7) y (206 ± 4), con porcentajes de recuperación del 83.7 % y 104.6 % respectivamente, por lo que el MOSA se puede aplicar con garantía. La aplicación del método de Youden antes y después del tratamiento de reducción, dio ordenada en el origen significativa en ambos casos: (170 ± 40) y (100 ± 40). Estos valores habrán de tenerse en cuenta para calcular la concentración de cromo en las muestras. Se observó que al aplicar el tratamiento de reducción, el valor del Youden disminuía, con lo que la señal que resultaba de la medida directa, sería debida a Cr(III) y a otras especies que desaparecían al aplicar el tratamiento de reducción. En los casos en que se observe este efecto, solo se podrá determinar el Cromo Total. El estudio de una muestra de agua mineralno proporcionó ninguna señal antes ni después del tratamiento de reducción, por lo que la muestra no contenía cromo. El análisis de esta muestra de agua mineral fortificada con 2.5 ìg/l de Cr(III) y 2.5 ìg/l Cr(VI), por el método quimioluminiscente propuesto y por el método de referencia de la difenilcarbazida [89], dio resultados similares (Tabla 51). Además los porcentajes de recuperación del Cromo adicionado a la muestra, fueron entre el 83-92 % utilizando el método QL, lo que reflejaba la exactitud del método. Tabla 51. Determinación de Cromo en una muestra de agua mineral. Método de Referencia Concentraciones (ìg/l) Método CL Concentraciones (ìg/l) Cr(III) Cr Total Cr(VI) Cr(III) Cr Total Cr(VI) Muestra (1.5±0.4) (1.7±0.2) - (1.56±0.05) (1.83±0.09) - Muestra fortificada (3.2±0.3) (6.1±0.3) (2.9±0.3) (3.7±0.1) (5.97±0.07) (2.3±0.2) La validación de la exactitud del método se completó al analizar la muestra de agua certificada (SRM © 1640), cuyo valor estimado fue (38.2 ± 1) ìg/l de Cromo (III) frente al valor certificado (38.6 ±1.6) ìg/l Cr(III).
- 142. REACCIÓN DEL LUMINOL 118 Conclusiones Se ha demostrado que para obtener resultados exactos en la especiación de Cromo, se han de tener en cuenta las siguientes consideraciones: § En la preparación de los reactivos hay que utilizar reactivos de pureza elevada (HCl(trace pure) / Na2CO3(p.a.)), para obtener blancos adecuados. § El Cr(III) tiene un comportamiento diferente antes y después de tratamiento de reducción debido a la ausencia o presencia de HCl en la muestra respectivamente. Las mezclas de Cr(III) y Cr(VI) dieron señales analíticas aditivas después del tratamiento. Se puede utilizar la misma curva de calibrado para la determinación de Cr(III) y Cr(VI) si se utiliza HCl en los patrones. § La conversión de Cr(VI) a Cr(III) es cuantitativa en las condiciones propuestas. A partir del estudio de interferencias, se observó que el Co(II) era un interferente fuerte si no se controla el pH y la concentración de EDTA. Sin embargo, sus señales son aditivas. § Para estudiar el efecto matriz, necesariamente se ha de utilizar el intervalo lineal de concentración. En las muestras analizadas no se encontró efecto matriz. Se propone utilizar el valor de la pendiente del MOSA para garantizar la ausencia de errores del método dada su robustez. El tratamiento de reducción utilizado para la determinación del Cr(VI), aumentaba la selectividad de la determinación de Cr(III). Para algunas muestras reales el método solo puede ser utilizado para la determinación de Cromo Total. § Siguiendo el protocolo establecido, la comparación con el método de referencia y el resultado del patrón de referencia certificado SRM © 1640, indican que los resultados obtenidos son exactos.
- 143. Capítulo 2. RESULTADOS OBTENIDOS Y DISCUSIÓN 119 2.2.1.2. INFLUENCIA DE LOS PROTOCOLOS DE ACONDICIONAMIENTO DE MUESTRAS DE AGUA EN LA DETECCIÓN QUIMIOLUMINISCENTE DE ELEMENTOS TRAZA. La conservación de las muestras es un problema analítico interesante debido a que la completa estabilidad de todos los constituyentes de la muestra es imposible de alcanzar. Después de la recogida de la muestra, los procedimientos de conservación solamente retrasan los cambios químicos y biológicos [89]. Los procesos de adsorción en la pared de los recipientes ocurren en muestras almacenadas sin acidificar y sin congelar. Las muestras pueden almacenarse no acidificadas pero congeladas para conservar la distribución original de las especies [90]. Independientemente de la temperatura de almacenamiento, muchos autores recomiendan la acidificación inmediatamente después del muestreo si se quieren analizar iones metálicos. El análisis inmediato para la determinación de metales en muestras de agua es siempre recomendable, o bien un tiempo de preservación corto de la muestra filtrada a baja temperatura (4ºC). En estos casos, la medida se realiza sin ningún otro paso de acondicionamiento. El análisis de agua embotellada se realiza también sin necesidad de ningún paso de acondicionamiento, sin embargo, el método más común de acondicionamiento para la determinación de metales en muestras de agua, es la adición de un ácido, como el HCl para corregir el pH hasta 2-3. Las muestras previamente filtradas se almacenan en frascos de plástico a 4ºC, el tiempo máximo recomendado es de 6 meses. El procedimiento de conservación a largo plazo utiliza la adición de HNO3. Las muestras se filtran, acidifican y guardan a 4ºC. Las botellas de polietileno se introducen en bolsas de plástico aluminizado. El tiempo de almacenaje puede durar incluso 2-3 años. Este es el protocolo de acondicionamiento que siguen los materiales de referencia certificados. La quimioluminiscencia proporciona sensibilidad y selectividad y acoplada a la inyección en flujo (FI), rapidez y reproducibilidad en la inyección de la muestra y mezclado de los reactivos, como se ha demostrado en el apartado anterior. Estos factores, junto al bajo coste comparado con la técnicas de AAS e ICP, la simplicidad y la posibilidad de realizar análisis in situ, hace a la determinación quimioluminiscente combinada con la inyección en flujo, extremadamente atractiva para la determinación de elementos traza en muestra de agua medioambientales. En la Tabla 52, se muestra una selección de las determinaciones quimioluminiscentes de iones metálicos en muestras de agua utilizando inyección en flujo descritas en la bibliografía [91-108]. La reacción más utilizada es la de oxidación del luminol con peróxido de hidrógeno en medio básico catalizada por iones metálicos. Para cuantificar Fe(II), Fe(III), Mn(II) y Cu(II), se requiere un paso de preconcentración, normalmente por cromatografía de intercambio iónico. Para complejar Mn(II) y Co(II), se han utilizado enmascarantes como EDTA, citrato sódico, 8-quinolinol o ácido fosfórico. En función de estas condiciones, los parámetros analíticos de los diferentes procedimientos se ven afectados.
- 144. REACCIÓN DEL LUMINOL 120 Tabla 52. Estudios de la determinación quimioluminiscente en flujo de iones metálicos en muestras de agua. Ref. Iones Reacción Pre- conc. Consideraciones del método Muestra de agua [91] Mn(II) luminol-H2O2 Si Muestra acidificada AcH-AcNH4 + pH=5.0. Conc Mn: 0.05-0.5 µg/L Marina [92] Fe(II) luminol-H2O2 Si H3PO4 en patrones/muestras pH=6. Conc Fe: 10-70 µg/L. - [93] Co(II) Cu(II) Fe(II) luminol-H2O2 Si Patrones 0.01mol/L HCl. Fase móvil ácido oxálico pH=4.2. Conc Co: 0.05-4 µg/L. Cu: 20-200 µg/L. Fe semicuantitativo. - [94] Fe(III) sulfoflavina- H2O2 Si Patrones 5⋅10-3 mol/L HCl. Muestra acidif. a pH=5.5. Conc Fe: 0.02-5.6 µg/L. Marina [95] Fe(III) luminol-H2O2 Si Muestra acidif. AcH-AcNH4 + a pH=3.0. Conc Fe(III): 0.005-0.1 µg/L. Oceánica [96] Cr(VI) luminol-hexa - cianoferrato(II) Si No acondicionamiento. Conc Cr: 0.04-1 µg/L. Residual [97] Co(II) Ácido gálico- H2O2 Si Muestras en ac fórmico/formato amónico a pH=3.6. Conc Co: 0.0006-1 µg/L. Marina de referencia [98] Mn(II) luminol-H2O2 Si Patrones en HCl 0.1mol/L. Conc Mn: 0.002-110 µg/L. Marina de referencia [99] Fe(II) Fe(III) luminol-O2. Si Patrones en HCl a pH=3.5. Conc Fe: 0.001-1 µg/L. suelo, río, lluvia, lago [100] Fe(II) Fe(III) luminol-O2. Si Patrones y muestras 0.1mol/L HCl. Conc Fe: 0.002-0.56 µg/L. Oceánica y marina [101] Fe(II) Total Fe Sulfoflavina- H2O2 Si Patrones en HCl. Muestras en tampón acetato pH=4.4. Conc Fe: 0.05-11 µg/L. Lluvia, río grifo, costa [102] Co(II) Fe(II) Fe(III) pirogalol-H2O2- NaOH, Si Patrones 0.01 mol/L en HCl purificado. Conc Co: 5-850 µg/L. Fe: - estuario [103] Cr(III) Fe(II) Co(II) luminol - H2O2- haluro No Con / sin EDTA. Conc Cr: 0.005-5.2 µg/L. Fe: 0.6-56 µg/L. Co: 0.06-59 µg/L. - [104] Co(II) Mn(II) luminol -IO4 - No Muestras H3PO4. Para deter Mn, 8-quinolinol, y para determ Co y Mn adic citrato. Conc Co: 0.01-12 µg/L. Mn: 0.02-9 µg/L. fresca y contaminada [82] Cr(III) luminol-H2O2 No Patrones y muestras 10-4 M EDTA y 3⋅10-4 M H3PO4. Disol. H2O2 10-3 M en EDTA. Conc Cr: 0.01-6 µg/L. mineral, potable, contaminada [105] Mn(II) TCNQ-O2-OH- EosinaY No No acid. Análisis inmediato. Disol. DDAB vesicular. Conc Mn: 4 – 4000 µg/L. potable [106] Fe(II) Mn(II) luminol-IO4 - No No acidif. o -fenantrolin activador determ. Fe. TEA agente complejante determ. Mn. Conc. Fe: 0.003-0.1 µg/L. Mn: 0.005-0.1 µg/L. Suelo, potable [86] Cr(III) luminol-H2O2 No H3PO4 en patrones/muestras pH=3.7. EDTA 10-4 M, portador carbonato 0.1 M y bromuro 0.3 M pH=10.8. Conc Cr: 0.5-500 µg/L. grifo [107] Cu(II) fenantrolina- H2O2 No Patrones en HCl a pH=3. Muestras a pH=2. Conc Cu: 0.006-0.45 µg/L. marina [88] Cr(III) luminol-H2O2 No EDTA 10-3 M en patrones/muestras. Conc Cr: 5.2-5200 µg/L natural [108] Cr(III) luminol-H2O2 No Portador EDTA- CO3 - pH 10.8. Muestras en condic ácidas. Conc Cr: 0.5-50 µg/L. río, grifo y potable
- 145. Capítulo 2. RESULTADOS OBTENIDOS Y DISCUSIÓN 121 En la bibliografía no existen referencias que estudien la influencia de las condiciones de acondicionamiento de las muestras en la señal quimioluminiscente. En este apartado, se estudia la señal quimioluminiscente directa producida por la reacción del luminol-peróxido de hidrógeno catalizada por iones metálicos como Cr(III), Co(II), Fe(II), Fe(III), Ni(II) y Mn(II) en función del protocolo de acondicionamiento de la muestra. Se han evaluado muestras sin acidificar o acidificadas con HCl o HNO3, tanto en estático como en flujo, y en presencia o ausencia de EDTA. No se ha utilizado ningún procedimiento de preconcentración. El montaje de inyección en flujo utilizado es el que se muestra en la Figura 17 Pág. 40 del Capítulo 1-Introducción. Más información acerca de las condiciones experimentales se refleja en el Apéndice 8 [78]. Señal QL de muestras no acidificadas (sin acondicionar) Se estudió inicialmente la señal quimioluminiscente de patrones preparados en agua ultrapura. La legislación especifica los niveles de concentración máximos permitidos para los metales en estudio en: 200 µg/L para Fe, 20 µg/L para Ni, 2 mg/L para Cu y 50 µg/L para Cr [40]. La composición de un agua natural en componentes traza puede ser descrita por el material de referencia certificado SRM © 1640 (Tabla 53). Tabla 53. Composición del material de referencia certificado SRM © 1640: agua dulce. Fracciones de masa certificadas Fracciones de masa de referencia El em ent o µµg/ Kg Ele men to µµg/ Kg Elemento µµg/Kg Aluminio 52.0 ± 1.5 Cobalto 20.28 ± 0.31 Cobre 34.3 ± 1.6 Antimonio 13.79 ± 0.42 Hierro 34.3 ± 1.6 Litio 27.89 ± 0.14 Arsénico 26.67 ± 0.41 Plomo 27.89 ± 0.14 Níquel 27.4 ± 0.8 Bario 148.0 ± 2.2 Manganeso 121.5 ± 1.1 Potasio 994 ± 27 Berilio 34.94 ± 0.41 Molibdeno 46.75 ± 0.26 Rubidio 2.00 ± 0.02 Boro 301.1 ± 6.1 Selenio 21.96 ± 0.51 Zinc 53.2 ± 1.1 Cadmio 22.79 ± 0.96 Plata 7.62 ± 0.25 Calcio 7.045 ± 0.089 Cromo 38.6 ± 1.6 Estroncio 124.2 ± 0.7 Magnesio 5.819 ± 0.056 Vanadio 12.99± 0.37 Silicio 4.73 ± 0.12 Sodio 29.35 ± 0.31 Talio* < 0.1 * Valor de Fracción de masa informativo.
- 146. REACCIÓN DEL LUMINOL 122 Se midió inicialmente la quimioluminiscencia utilizando el sistema de inyección en flujo. Ni(II) y Mn(II) dieron señales QL similares a las del blanco. Fe(II) y Fe(III) solo dieron señales significativas a niveles superiores a los límites legislados (0.2 mg/l), por lo que sería necesario utilizar un paso de preconcentración para aplicar el método a la determinación de estos metales. Cr(III), Co(II) y Cu(II) dieron señales significativas para su cuantificación. Las curvas de calibrado doble-logarítmicas y en los intervalos lineales de concentración, en las diferentes condiciones experimentales ensayadas, se muestran en la Tabla 54. Se utilizó la altura de pico como señal analítica. El intervalo lineal de concentraciones fue: para Cr(III) 3-15 µg/L, para Co 2-8 µg/L, y para Cu, hasta 20 µg/L. En ausencia de EDTA, los límites de detección calculados para una relación señal/ruido de 3, estuvieron entre 0.5-2 µg/L. Estos valores son similares a los que se obtienen utilizando absorción atómica electrotérmica para elementos traza [109]. También se estudió el uso de EDTA como agente complejante de interferentes, añadiéndolo a la muestra o al portador (tampón o KOH), de forma que el complejo metal- EDTA se formaba antes o durante el sistema de flujo respectivamente. En presencia de EDTA, se obtiene que la sensibilidad del Co(II) disminuye drásticamente. Para el Cu(II), la señal desapareció completamente. La sensibilidad del Cr(III) disminuyó al adicionar EDTA 0.01 M al portador, como puede verse en la Tabla 54. Al utilizar EDTA 5· 10-3 M en el portador (tampón), se observó que la sensibilidad para el Cr(III) era similar a la obtenida en ausencia de EDTA, por lo que en estas condiciones, la sensibilidad se mantenía, y la reacción era además selectiva para Cr(III). Para el procedimiento en estático, las curvas de calibrado para Cromo y Cobalto en presencia de EDTA 3· 10-3 M, se muestran en la Tabla 55. Como puede verse, la sensibilidad del Cr resultó 3 veces superior a la del Co. Al adaptar el montaje FIA al fluorímetro utilizado para las medidas en estático, la curva de calibrado que se obtuvo para Cr fue: y = (0.07 ± 0.05) + (0.30 ± 0.02)· C (r2 = 0.997, sy/x = 0.07), por lo que la pérdida de sensibilidad del procedimiento en flujo respecto al procedimiento en estático, fue del orden del 60%.
- 147. Capítulo 2. RESULTADOS OBTENIDOS Y DISCUSIÓN 123 Tabla 54. Parámetros analíticos para la determinación de Cr(III), Co(II) y Cu(II) en muestras no acidificadas. Procedimiento en flujo. Sin EDTA EDTA en la muestra EDTA en el portador de KOH (a) EDTA en el portador de tampón(b) Conc EDTA (M) - 1⋅⋅10-2 5⋅⋅10-3 1⋅⋅10-2 5⋅⋅10-3 1⋅⋅10-2 5⋅⋅10-3 Doble logarítmica Cr(III) log a 1.82±0.06 2.4±1.7 1.5±0.6 2.2±1.5 1.84±0.03 1.9±0.7 2.32±0.03 a 66±9 247±54 32±4 163±34 69±5 99±6 211±16 b 1.32±0.06 0.4±0.1 1.14±0.06 0.65±0.09 1.16±0.03 0.95±0.03 0.92±0.04 r2 0.977 0.7086 0.9846 0.8284 0.9907 0.9926 0.985 n, sy/x 4, 0.0135 3, 0.208 3, 0.1090 4, 0.202 4, 0.0763 3, 0.0625 4, 0.1466 Co(II) log a 3.0±0.1 -0.6±1.2 -0.7±1.7 0.44±0.16 0.8±0.4 1.5±0.7 1.0±0.3 a 1009±121 0.27±0.06 0.21±0.02 2.7±1.0 6.8±2.6 32±5 10±2 b 0.70±0.08 1.53±0.05 1.51±0.02 1.08±0.08 0.82±0.08 0.62±0.03 0.86±0.04 r2 0.8941 0.9919 0.9442 0.9502 0.9112 0.9821 0.9825 n, sy/x 4, 0.1373 3, 0.0887 3, 0.162 4, 0.1406 4, 0.1466 3, 0.053 4, 0.065 Cu(II) log a 2.45±0.04 - - - - - - a 265±34 - - - - - - b 0.69±0.04 - - - - - - r2 0.9809 - - - - - - n, syx 4, 0.0682 - - - - - - Intervalo Lineal Cr(III) b0 108±77 115±50 -9±17 45±49 -17±32 30±19 6±44 b1 155±8 51±6 47±2 69±5 105±3 85±2 176±4 r2 0.9647 0.8943 0.9816 0.9327 0.9868 0.9937 0.9913 n, sy/x 5, 172.3 4, 111.6 4, 38.54 5, 110.62 5, 72.47 4, 42.62 5, 97.98 Co(II) b0 251±190 2.4±23 22±13 -19±33 19±18 87±31 37±17 b1 553±39 3.1±0.3 1.8±0.2 4.2±0.3 2.71±0.15 4.1±0.3 4.64±0.12 r2 0.9396 0.9166 0.9606 0.949 0.9604 0.9648 0.9899 n, sy/x 5, 425.76 3, 43.23 3, 103.35 5, 74.24 5, 41.79 4, 66.78 5, 35.56 Cu(II) b0 98±38 - - - - - - b1 111±3 - - - - - - r2 0.9972 - - - - - - n, sy/x 5, 58.61 - - - - - - (a) 0.02 mol/L KOH (b) 0.3 mol/L CO3 2- /HCO3 - (pH=10.8)
- 148. REACCIÓN DEL LUMINOL 124 Tabla 55. Parámetros analíticos de las curvas de calibrado de patrones no acidificados utilizando el procedimiento en estático. Metal Metal adicionado bo ±± sb0 b1 ±± sb1 syx R 2 n Cr - 0.3 ± 0.6 0.75 ± 0.05 1.2 0.97 8 Co - -0.2 ± 0.7 0.24 ± 0.01 0.9 0.99 5 Co - -0.2 ± 1 0.27 ± 0.01 3 0.99 8 Co - -0.2 ± 0.6 0.26 ± 0.01 0.8 0.996 4 Cr 80 µg/L Co 19 ± 3 0.85 ± 0.11 3 0.98 3 Cr 100 µg/L Co 30.5 ± 0.3 0.77 ± 0.02 0.3 0.999 3 Cr 200 µg/L Co 53.1 ± 0.8 0.68 ± 0.16 3 0.95 3 Co 20 µg/L Cr 16 ± 2 0.26 ± 0.02 3 0.98 5 Co 20 µg/L Cr 15 ± 2 0.24 ± 0.04 2 0.97 3 Co 40 µg/L Cr 39 ± 3 0.23 ± 0.04 3 0.94 4 Señal QL de muestras acondicionadas con HCl 5· 10-3 M Para el procedimiento en flujo, se evaluó la etapa de calibración en condiciones ácidas con la adición de HCl 5· 10-3 M. Se estudió el efecto de la presencia de EDTA en el portador o en la muestra obteniéndose un comportamiento similar al obtenido en muestras no acidificadas. En la Tabla 56 se muestran los parámetros de calibración para los diferentes modelos de calibración obtenidos en las diferentes condiciones experimentales. Las sensibilidades alcanzadas fueron similares a las obtenidas en las condiciones de no acidificación, obteniéndose límites de detección sin EDTA similares a los del apartado anterior. En presencia de EDTA, la sensibilidad del Cr(III) se mantiene igual que sin acondicionar; para el Cu(II) la señal quimioluminiscente desaparece; y para el Co(II) la sensibilidad aumenta ligeramente respecto a los valores sin acondicionamiento de HCl, alcanzándose límites de detección del orden de 50 µg/l. Para el procedimiento en estático, las sensibilidades obtenidas para Cr y Co en presencia de EDTA fueron 0.898 y 0.35 respectivamente. Estos valores fueron similares a los establecidos en la Tabla 55, para muestras no acondicionadas.
- 149. Capítulo 2. RESULTADOS OBTENIDOS Y DISCUSIÓN 125 Tabla 56. Parámetros analíticos para la determinación de Cr(III), Co(II) y Cu(II) en muestras acidificadas con HCl 5· 10 -3 M. Procedimiento en flujo. Sin EDTA EDTA en la muestra EDTA en portador- KOH(a) EDTA en portador- Tampón(b) Conc EDTA (M) - 1⋅⋅10-2 5⋅⋅10-3 5⋅⋅10-3 1⋅⋅10-2 5⋅⋅10-3 Doble Logarítmica Cr(III) log a 2.31±0.02 1.2±0.3 1.3±0.5 2.1±1.1 2.1±0.9 2.5±1.6 a 203±8 18±2 22±3 129±13 144±9 334±39 b 0.88±0.02 1.43±0.05 1.50±0.06 0.89±0.04 0.89±0.03 0.78±0.05 r2 0.9958 0.9913 0.9870 0.9747 0.9914 0.9574 n, s y/x 4, 0.0383 3, 0.217 3, 0.1312 4, 0.0979 4, 0.0628 4, 0.1122 Co(II) log a 2.99±0.03 1.0±0.7 -(0.8±1.4) 0.7±0.6 0.9±0.2 1.4±0.3 a 976±78 11±5 0.17±0.04 6±4 9±1 25±2 b 0.81±0.05 1.04±0.09 1.63±0.06 1.14±0.15 0.88±0.02 0.718±0.017 r2 0.9621 0.9442 0.9914 0.8595 0.9948 0.9939 n, s y/x 4, 0.0916 3, 0.162 3,0.0972 4, 0.263 4, 0.041 4, 0.0319 Cu(II) log a 3.06±0.04 - - - - - a 1276±146 - - - - - b 0.30±0.03 - - - - - r2 0.8783 - - - - - n, s y/x 4, 0.0744 - - - - - Intervalo Lineal Cr(III) b0 71±30 -15±22 -80±56 69±48 39±19 153±137 b1 144±3 54±3 90±6 93±5 106±2 184±14 r2 0.9938 0.9773 0.9528 0.9629 0.9963 0.9368 n, s y/x 5, 68.16 4, 49.72 4, 126.41 5, 108.95 4, 42.82 5, 275.59 Co(II) b0 374±184 79±55 -10±26 83±41 31±9 66±12 b1 613±37 11.1±0.8 3.4±0.4 9.6±0.6 5.33±0.13 6.55±0.18 r2 0.85354 0.9606 0.9095 0.9701 0.9955 0.9953 n, s y/x 5, 411.12 3, 103.35 3, 49.57 3, 77.97 3, 16.56 3, 20.27 Cu(II) b0 483±389 - - - - - b1 131±27 - - - - - r2 0.8185 - - - - - n, s y/x 3, 606.64 - - - - - (a) 0.02 mol/L KOH (b) 0.3 mol/L CO3 2- /HCO3 - (pH=10.8) Señal QL de muestras acondicionadas con HNO3 0.5 M Se estudió la emisión quimioluminiscente en condiciones drásticas (HNO3 0.5M). En estas condiciones y utilizando el procedimiento en flujo, las curvas de calibrado obtenidas sin EDTA para Cr (III), Co(II) y Cu(II), dieron sensibilidades bastante peores que en las anteriores condiciones de acondicionamiento (ver Tabla 57). Sin embargo, la pendiente para el Cr(III), fue similar a la obtenida en patrones no acidificados o
- 150. REACCIÓN DEL LUMINOL 126 acidificados con HCl, en presencia de EDTA 0.01M. Los límites de detección en ausencia de EDTA calculados para una relación señal/ruido de 3, fueron: 3, 3.5 y 10 µg/L para Cr(III), Co(II) y Cu(II) respectivamente. Tabla 57. Parámetros analíticos para la determinación de Cr(III), Co(II) y Cu(II) en muestras acidificadas con HNO3 0.5 M. Procedimiento en flujo. Doble Logarítmico Intervalo Lineal Cr(III) Co(II) Cu(II) Cr(III) Co(II) Cu(II) log a 2.6 ± 1.6 2.8 ± 1.6 2.6 ± 1.6 b0 513 ± 29 484 ± 48 579 ± 15 a 455 ± 44 678 ± 43 367 ± 38 b1 60 ± 3 130 ± 10 9.1 ± 0.5 b 0.39 ± 0.05 0.35 ± 0.04 0.26 ± 0.03 r2 0.9619 0.9296 0.9717 r2 0.8799 0.8816 0.9143 n, sy/x 5, 66.33 5, 108.63 4, 31.80 n, sy/x 4, 0.0932 4, 0.0733 3, 0.040 Para el procedimiento en estático, se requirió la neutralización de la muestra. Los mejores resultados se obtuvieron neutralizando con Na2CO3 0.2272M para una dilución 7/10 de las muestras. Las señales QL para Cr y Co en estas condiciones, se muestran en la Figura 46. Las curvas de calibrado para Cr y Co fueron diferentes a las obtenidas en las condiciones anteriores: Cr (III): y = (-0.4 ± 0.2) + (0.38 ± 0.01)⋅CCr (r2 =0.99, syx=1.5) Co (II): y = (-0.2 ± 0.2) + (0.54 ± 0.01)⋅CCo (r2 =0.99, syx=0.1) Figura 46. Señales quimioluminiscentes para Cr, Co y Material Estándar de Referencia (SRM © 1640). Procedimiento en estático. 0 10 20 30 40 50 SeñalQuimioluminiscente Cr (III) (ng/ml) Co(II) (ng/ml) SRM 0 15 27 45 65 85 20 60 80 0
- 151. Capítulo 2. RESULTADOS OBTENIDOS Y DISCUSIÓN 127 Aplicabilidad La señal quimioluminiscente variaba en función del protocolo de acondicionamiento de la muestra y de la presencia de agente enmascarante, aunque esta influencia era mínima para el Cr(III). Añadiendo EDTA se obtuvo especificidad para la determinación de Cromo, sin que se requiriera neutralización al utilizar el procedimiento en flujo. Según las condiciones experimentales seleccionadas, la reacción resultó selectiva para la determinación de Cr(III), o bien se puede evaluar la contribución total de Cr, Co y Cu. En la Figura 47, se muestran las pendientes obtenidas para cada uno de los metales estudiados en las diferentes condiciones ensayadas utilizando el procedimiento en flujo. El método en estático requirió la adición de Na2CO3 al acondicionar las muestras en condiciones ácidas drásticas (HNO3 0.5M). Figura 47. Pendiente de la curva de calibrado en el intervalo lineal en las diferentes condiciones experimentales. 1: Sin acondicionar; 2: HCl 5· 10-3 ; 3: HNO3 0.5; 4: EDTA 10-2 ; 5: EDTA 1· 10-2 , HCl 5· 10-3 ; 6: EDTA 5· 10-3 ; 7: EDTA 5· 10-3 , HCl 5· 10-3 ; 8: EDTA 10-2 (portador KOH); 9: EDTA 10-2 (portador tampón); 10: EDTA 5· 10-3 (portador KOH); 11: EDTA 5· 10-3 (portador tampón); 12: EDTA 10-2 (portador tampón), HCl 5· 10-3 ; 13: EDTA 5· 10-3 (portador KOH), HCl 5· 10-3 ; 14: EDTA 5· 10-3 (portador tampón), HCl 5· 10-3 . Concentración: mol/L. Para estudiar la señal quimioluminiscente total, se llevaron a cabo varias experiencias, con la intención de evaluar la aditividad de las señales en las diferentes condiciones estudiadas. Se estudiaron mezclas binarias y terciarias de los tres metales almacenados en condiciones ácidas y no ácidas (ver parte experimental en el Apéndice 8, [78]). Las mezclas dieron porcentajes de recuperación de señal próximos al 100% (Figura 48), demostrándose así la aditividad entre las señales de los tres metales tanto en estático (mezclas s1, s2, s3, s4) como en flujo (mezclas s5, s6, s7, s8, s9, s10, s11, s12, s13, s14, s15, s16). 0 100 200 300 400 500 600 700 800 1 2 3 4 5 6 7 8 9 10 11 12 13 14 Acondicionamiento de la muestra b(L/mg) Cr Co Cu
- 152. REACCIÓN DEL LUMINOL 128 Figura 48. Porcentajes de recuperación de señal para diferentes mezclas de metales. Además, para el procedimiento en estático, se realizó un diseño experimental 52 de mezclas de patrones. El test t demostró que las pendientes de las curvas de calibrado de Cr(III) obtenidas en presencia de diferentes concentraciones de Co(II) eran similares con un nivel de confianza del 95%. Los mismos resultados se obtuvieron al aplicar el test t para la comparación de curvas de calibrado de Co(II) en presencia de Cr(III). Algunas de estas ecuaciones de calibración se muestran en la Tabla 55. Los valores de pendiente para las mezclas son similares a los del elemento traza solo. Por tanto, la señal total puede considerarse como la suma de las señales individuales de Cr y Co. Para las mezclas de calibración utilizando el procedimiento en flujo, se hizo un estudio similar, y en todos los casos, el test t demostró que no había diferencias significativas entre las pendientes de calibración a un nivel de confianza del 95%. Se estudió el material de referencia certificado (SRM © 1640) de agua dulce conservado en condiciones de HNO3 0.5M. Los contenidos en metales se han dado previamente en la Tabla 53. En la Figura 46, aparece la señal analítica obtenida para el material de referencia certificado utilizando el procedimiento en estático. Al igual que en los patrones, se le añadió Na2CO3. El porcentaje de recuperación respecto a una mezcla de Cr y Co preparada en las mismas condiciones que el material de referencia certificado fue cercano al 100% (Figura 49), por lo que únicamente estos dos elementos contribuían a la señal analítica total. Principalmente, la señal se debía al Cromo, porque la concentración del Co considerando una dilución 7/10 era cercana al límite de detección. Utilizando el procedimiento en flujo, se podía cuantificar Cr añadiendo EDTA 0.01 M, o bien la contribución total de Cr, Co y Co en ausencia del complejante. Para determinar Cr, se podía utilizar patrones en condiciones ácidas o no ácidas para procesar muestras acondicionadas en diferentes condiciones. Esto puede observarse a partir de los 0 20 40 60 80 100 120 140 s-1 s-2 s-3 s-4 s-5 s-6 s-7 s-8 s-9 s-10 s-11 s-12 s-13 s-14 s-15 s-16 Mezclas Porcentajederecuperación Cu Co Cr
- 153. Capítulo 2. RESULTADOS OBTENIDOS Y DISCUSIÓN 129 valores de sensibilidad y de los resultados obtenidos para el SRM © 1640 mostrados en la Figura 49. Sin EDTA, los patrones y muestras han de estar en las mismas condiciones para obtener resultados exactos. En la Figura 49 se demuestra que se obtuvieron los mismos resultados para el SRM © 1640 y para una mezcla de Cr, Co y Cu preparada en HNO3. Figura 49. Porcentajes de recuperación del SRM © 1640 respecto a mezcla de patrones en diferentes condiciones experimentales. Conclusiones Se estudió la señal quimioluminiscente para diferentes protocolos de acondicionamiento de muestras. Además se hizo un estudio paralelo de la selectividad. Se calcularon los modelos de calibración para cada uno de los protocolos de acondicionamiento. Los estudios de recuperación mostraron la aditividad de las señales para diferentes condiciones. La selectividad, exactitud y precisión se evaluaron utilizando un material de referencia certificado. Se propuso una guía para la determinación de metales basada en la señal quimioluminiscente. El procedimiento de acondicionamiento de muestras y patrones se elegiría de acuerdo con la estrategia de muestreo, los objetivos de análisis (uni- o multielemental) y las necesidades de acondicionamiento. Por tanto, los métodos quimioluminiscente en estático o en flujo, pudieron aplicarse satisfactoriamente al análisis de aguas naturales acondicionadas en diferentes condiciones. En presencia de EDTA, fue posible la determinación selectiva de Cr(III) en aguas naturales. La determinación de Cr(III) no dependió del protocolo de acondicionamiento cuando se trabajó en flujo. Por tanto, se pueden utilizar patrones preparados en agua para procesar muestras preparadas en diferentes condiciones si se utiliza como portador EDTA 0.01 M. Al utilizar el método en estático, el pH de la muestra debe controlarse añadiendo Na2CO3. 0 20 40 60 80 100 120 140 SinEDTA EDTA EDTA EDTA SinEDTA Porcentajederecuperación(%) FI no acidif. FI adic.-HCl FI adic.-HNO3 FI adic.-HNO3 estático adic.-HNO3
- 154. REACCIÓN DEL LUMINOL 130 2.2.1.3. CALIBRACIÓN MULTIVARIADA APLICADA A LA DETERMINACIÓN QUIMIOLUMINISCENTE SIMULTÁNEA DE COBALTO Y CROMO. En la bibliografía se encuentran numerosos métodos analíticos propuestos para la determinación de cromo y/o cobalto, como la electroforesis capilar [110], NAA [111], XRF [112-113], AAS[114-115], ICP-AES [116], o ICP-MS [112,117]. Se han propuesto diferentes estrategias para la determinación selectiva o simultánea de iones metálicos utilizando la reacción quimioluminiscente del luminol. En los sistemas de inyección en flujo, se han utilizado pre-tratamientos o procedimientos de separación. Se ha publicado el uso de agentes enmascarantes [104], separadores de membrana [118] o resinas de intercambio iónico [119]. Para la determinación simultánea de varios metales se ha propuesto la cromatografía iónica con detección quimioluminiscente [120,81]. Sin embargo, en este caso, la limitación es la incompatibilidad de la fase móvil con las condiciones de reacción requeridas para la reacción postcolumna [93]. Se han resuelto mezclas de compuestos orgánicos [121-122] y de iones metálicos [123-124] por calibración multivariada y/o la técnica de eliminación de interferentes. El método de mínimos cuadrados parciales (PLS) se ha aplicado a la determinación simultánea de mezclas de compuestos en muestras de agua. En este apartado, se estudia un modelo de mínimos cuadrados parciales (PLS) para la determinación simultánea de Cr(III) y Co(II) en muestras de agua, registrando en estático la curva cinética, y construyendo diferentes modelos a partir del conjunto de medidas señal-tiempo. La capacidad de predicción se ha evaluado utilizando disoluciones patrón, y el modelo ha sido validado en la determinación de concentraciones de Cr(III) y Co(II) en un material de referencia certificado de agua natural. Más información acerca de las condiciones experimentales, se refleja en el Apéndice 9 [125]. Estudio de la señal quimioluminiscente Cuando se trabaja en estático, la emisión quimioluminiscente del sistema luminol/H2O2 describe una curva de respuesta Intensidad frente al Tiempo de primer orden [126]: formación y destrucción del emisor de luz (3-aminoftalato en este caso). La velocidad de reacción aumenta con la concentración de diferentes iones metálicos. La curva de respuesta depende de factores experimentales como el pH o la velocidad de mezclado [127], pero su influencia se evitó manteniéndolos constantes. También depende del metal presente en la disolución, siendo Cr y Co los metales que contribuyen principalmente a la señal quimioluminiscente en las condiciones estudiadas, tal como se demuestra en el capítulo anterior. La contribución de otras especies se consideró despreciable.
- 155. Capítulo 2. RESULTADOS OBTENIDOS Y DISCUSIÓN 131 En la Figura 50, se muestran los perfiles Intensidad-Tiempo para patrones de 20, 40, 60 y 80 ìg/l de Cromo y las mismas concentraciones de Cobalto. Son perfiles normalizados, en los que se da el valor de 1 al máximo de emisión. Se observó que para ambos metales el perfil de formación del emisor quimioluminiscente (aumento de la señal QL) fue similar, por lo que en los modelos descritos posteriormente, se estudió la posibilidad de eliminar del conjunto de calibración las 15 primeras variables, correspondientes a esta región de formación del emisor quimioluminiscente. Por otra parte, el perfil de destrucción del emisor (disminución de la señal QL) fue diferente en función del metal estudiado: para el Cr la destrucción era más rápida que para el Co. Los perfiles obtenidos fueron robustos, siendo el comportamiento descrito independiente de la concentración del metal en la disolución. Figura 50. Perfiles normalizados Intensidad-Tiempo para Cr y Co. Evaluación de los modelos de calibración Se estudiaron 2 opciones como variables predictoras: el registro completo (62 variables) o la región de destrucción (46 variables). Se ensayaron los modelos PLS-1 y PLS-2. Además, los modelos se obtuvieron trabajando en dos condiciones de calibración diferentes: utilizando el intervalo lineal de concentraciones (0-20 ìg/l Cr, 0-80 ìg/l Co) para el cual se evaluó la aditividad de las señales producidas por ambos metales; o utilizando intervalos de concentraciones en que la respuesta no era lineal (0-80 ìg/l Cr, 0- 80 ìg/l Co). 0 0.2 0.4 0.6 0.8 1 0 1 2 3 4 5 6 Tiempo (s) SeñalQLnormalizada Cr Co 15 variables 46 variables Región de formación Región de destrucción
- 156. REACCIÓN DEL LUMINOL 132 S Respuesta lineal Se utilizaron dos aproximaciones para evaluar la aditividad de la señal: Aproximación empírica-teórica: El conjunto de calibración se compone de registros bicomponentes teóricos, que se obtuvieron a partir de los registros unicomponentes experimentales. Se representó la señal frente a la concentración del metal para cada tiempo, deduciéndose valores de pendientes en función del tiempo para los dos metales estudiados. Los registros bicomponentes teóricos (pendiente de la mezcla-tiempo), se dedujeron de la suma de las pendientes de los dos metales en cada tiempo. A la suma de las pendientes se añadió un error heterocedástico aleatorio simulado, de la misma magnitud que la reproducibilidad de la señal. En la Figura 51, se muestran los perfiles descritos. Figura 51. Perfiles experimentales Pendiente-Tiempo para Cr y Co, y perfil teórico de la mezcla incluyendo el error heterocedástico. Aproximación empírica: El conjunto de calibración se compone de registros unicomponentes experimentales. En ambas aproximaciones, se obtuvieron modelos de PLS-1 y PLS-2 con 61 y 46 variables, obteniéndose porcentajes de varianza explicada cercanos al 99% en la mayoría de los casos, utilizando modelos de 2 componentes principales, y con errores de predicción bajos. Los datos obtenidos en cada caso se muestran en la Tabla 58. 0.0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9 1.0 0 2 4 6 Tiempo (s) 0 1 2 3 4 5 6 7 8 9 10 Pendiente Co Pendiente Cr Mezcla + Ruido Pendiente Señalmezcla
- 157. Capítulo 2. RESULTADOS OBTENIDOS Y DISCUSIÓN 133 Tabla 58. Modelos PLS en el intervalo lineal. % Varianza Explicada RMSEP calibración RMSEP validación Variables VL* Bloque X Bloque Y Cr Bloque Y Co Cr Co Cr Co 1-61 2 98.70 99.88 99.90 0.8 2.7 1.7 4.9 PLS-1 16-61 2 99.92 94.22 99.71 5.9 7.6 6.4 11.4 1-61 2 98.70 99.89 0.8 2.7 1.7 4.9 Aprox. Empírica- Teórica PLS-2 16-61 2 99.92 99.36 6.0 7.5 6.7 11.2 1-61 2 99.98 99.98 99.99 1.5 4.3 2.7 7.7 PLS-1 16-61 2 99.99 95.65 99.58 6.2 9.3 7.7 12.5 1-61 2 99.98 99.99 1.5 4.3 2.7 7.7 Aprox. Empírica PLS-2 16-61 2 99.99 99.35 6.2 9.3 7.7 12.4 *VL: Variables latentes. En ambas aproximaciones, se confirmó la aditividad de las señales quimioluminiscentes de Cr y Co, libre de efectos sinérgicos. La robustez de los modelos obtenidos se demostró por la similitud entre los errores de predicción utilizando el conjunto de calibración o el de validación. Los errores de predicción fueron menores al utilizar las 61 variables. Con estos resultados, se concluyó que la predicción de mezclas de Cr y Co es posible cuando se trabaja en el intervalo lineal de concentraciones, utilizando como conjunto de calibración los registros de disoluciones unicomponentes o los registros bicomponentes simulados. S Respuesta no lineal Se partió de registros unicomponentes y bicomponentes de Cr y Co en el intervalo de concentraciones de 0-80 ìg/l. Se aplicó PLS-1 y PLS-2 a este conjunto de calibración, tomando como Bloque X la señal QL a cada tiempo (61 o 46 variables), y como Bloque Y las concentraciones de Cr y Co. El gráfico de las puntuaciones para las dos primeras componentes principales del modelo (Figura 52 B), presenta una distribución similar al diseño experimental (Figura 52 A), pero la forma cuadrada se pierde un poco debido a que el primer factor también describe la contribución no lineal. Como puede observarse, el primer factor está relacionado con la concentración de Cromo, y el segundo factor está relacionado con la concentración de Cobalto.
- 158. REACCIÓN DEL LUMINOL 134 Figura 52. A) Diseño experimental 52 . B) Gráfico de puntuaciones con las dos primeras CPs. El hecho de tener una relación señal-concentración no lineal, llevó a la obtención de modelos de 4 componentes principales cuando se trabajó con las 61 variables, y modelos de 3 componentes principales cuando se trabajó con 46 variables, tal como puede observarse en la Tabla 59. Tabla 59. Modelos de PLS en el intervalo no lineal. PLS-1 PLS-2 Variables 1-61 16-61 1-61 16-61 Variables latentes (VL) 4 3 4 3 Bloque X 99.93 99.94 99.93 99.92 Bloque Y Cr 92.81 95.97 % Varianza Explicada Bloque Y Co 78.62 96.24 85.14 95.46 Cr 7.6 5.7 7.5 6.5RMSEP Calibración Co 13.1 5.5 13.5 5.5 Cr 10.6 7.4 10.5 7.4RMSEP Validación Co 18.0 7.5 17.8 8.4 Los porcentajes de varianza explicada fueron mejores al utilizar el modelo de 46 variables, proporcionando asimismo menores errores de predicción. La selección previa de variables con información selectiva proporciona modelos de PLS con menor número de factores y una capacidad de predicción similar. Los errores de predicción para los conjuntos de calibración y validación fueron similares, remarcando la robustez del modelo. Con esto, se concluyó que la determinación simultánea de Cromo y Cobalto era posible en el intervalo de concentraciones estudiado. 0 20 40 60 80 0 20 40 60 80 Concentración Cr (µ g/l) ConcentraciónCo(g/l) A) 00 40 04 44 -3 -2 -1 0 1 2 3 -100 -50 0 50 100 CP1 CP2 Cr Co B) 00 04 44 40
- 159. Capítulo 2. RESULTADOS OBTENIDOS Y DISCUSIÓN 135 Validación Para validar el modelo, se utilizó un material de referencia certificado de agua dulce SRM © 1640, que contenía además de Cromo y Cobalto, otras especies que se encuentran habitualmente en muestras de agua (ver Tabla 53, Pág. 121). El modelo de calibración se construyó a partir de los registros quimioluminiscentes de disoluciones unicomponentes, preparadas en las mismas condiciones que el material de referencia certificado (en ácido nítrico). Las características para los diferentes modelos obtenidos, se muestran en la Tabla 60. Tabla 60. Modelos de PLS en condiciones ácidas. % Varianza Explicada RMSEP Calibración RMSEP Validación Modelo Variables VL Bloque X Bloque Y-Cr Bloque Y-Co Cr Co Cr Co 1-61 4 99.94 99.94 99.80 0.7 1.2 2.6 4.6 PLS-1 16-61 3 99.93 99.94 96.63 0.7 5.1 4.1 10.3 1-61 4 99.94 99.47 1.7 2.3 3.4 5.6 PLS-2 16-61 3 99.94 99.47 0.9 5.6 4.4 10.1 Los porcentajes de varianza explicada fueron cercanos al 100%, tanto utilizando modelos de PLS-1 como de PLS-2, y para los modelos con 46 variables, el número de variables latentes del modelo fue menor que utilizando las 61 variables, pero los errores de predicción tanto de calibración como de validación fueron mayores. Para el Co, es preferible utilizar elmodelo de 61 variables. El estudio de mezclas de Cr/Co en estas condiciones, dio las rectas de valor predicho frente a valor real que se muestran en la Tabla 61. En cualquier caso, la pendiente resultó estadísticamente igual a 1 y la ordenada estadísticamente igual a 0, lo que indicaba que los modelos propuestos eran buenos para la determinación simultánea de Cr y Co en condiciones ácidas. Tabla 61. Rectas de regresión lineal Valor Predicho frente a Valor Real. Predicho frente a medido Modelo Analito a b R2 sy/x PLS-1 Cr 0 ± 2 0.98 ± 0.07 1.0 3.3 Co -2 ± 6 0.9 ± 0.2 0.9 8.8 PLS-2 Cr 0 ± 2 0.98 ± 0.07 1.0 3.4 Co 0 ± 4 0.94 ± 0.15 0.9 6.5 La concentración predicha para Cr y Co en el material de referencia certificado utilizando los modelos propuestos (Tabla 62), fue comparada con los valores certificados mediante un test t con un nivel de probabilidad del 95%, obteniéndose resultados
- 160. REACCIÓN DEL LUMINOL 136 similares al utilizar el registro completo (61 variables), quedando la exactitud del método validada. Tabla 62. Predicción para el material de referencia certificado (µg/L, n=5). Concentración (µµg/L) t calculado* Variables VL Cr Co Cr Co Referencia - - 38.6 ± 1.6 20.28 ± 0.31 - - PLS-1 1-61 4 32 ± 7 21 ± 7 -2.4 0.3 16-61 4 (35 ± 2) (11 ± 6) (5.7) (-3.1) PLS-2 1-61 3 31 ± 7 25 ± 9 -2.1 1.0 16-61 3 (36 ± 2) (12 ± 5) 2.5 (-3.8) *t tabulado (95%, 4) = 2.776. Este material de referencia certificado sirvió además para demostrar que la contribución a la señal QL viene dada principalmente por los dos metales estudiados, siendo despreciable la interferencia de los demás metales presentes, como ha sido demostrado en el capítulo anterior. Con este estudio, se demostró la robustez de las señales quimioluminiscentes y se propuso y validó un método capaz de predecir concentraciones de Cromo y Cobalto en muestras de agua evitando la interferencia de otros elementos. Conclusiones En este capítulo, se utilizó el PLS para modelizar la respuesta quimioluminiscente en función del tiempo. Con ello, se hizo posible la determinación simultánea y selectiva de dos metales (cromo y cobalto) en muestras de agua. Se ensayaron diferentes diseños experimentales para evaluar la aditividad entre las señales y la relación no-lineal entre la respuesta y la concentración. El conjunto de calibración se seleccionó en base a dos criterios: disoluciones de patrones unicomponentes y/o bicomponentes y pendiente calculada a partir de los diferentes calibrados lineales univariados. También se estudió la selección de las variables predictoras. En todos los casos se obtuvieron buenos resultados, aunque los mejores resultados correspondieron a la utilización del registro completo (aumento y disminución de la señal quimioluminiscente). El método se validó utilizando mezclas de patrones y un material de referencia certificado. Este método presentó las ventajas de su bajo coste, respuesta rápida y no requiere personal cualificado. Estas características son muy útiles para la monitorización de Cr y Co en análisis de aguas.
- 161. Capítulo 2. RESULTADOS OBTENIDOS Y DISCUSIÓN 137 Las novedades que aporta esta publicación frente a otras publicaciones existentes consisten en que: § Se ha demostrado la robustez de los perfiles intensidad-tiempo. § Es la primera aplicación de estos perfiles intensidad-tiempo en calibración multivariada para el análisis quimioluminiscente simultáneo de dos especies. Otras publicaciones utilizan registros intensidad-longitud de onda obtenidos a partir de dispositivos especiales (DDC) para determinar un analito [128-129]. § Se han validado los resultados con un material de referencia certificado.
- 162. REACCIÓN DEL LUMINOL 138 1.2. DETERMINACIÓN DE NITRÓGENO ORGÁNICO AMINO Y AMONIACAL. El Nitrógeno es un nutriente clave en aguas naturales, pero los vertidos de nitrógeno en exceso puede causar procesos de eutrofización de las aguas. Las especies dominantes de nitrógeno en las aguas son: el nitrógeno inorgánico disuelto (amonio, nitrito y nitrato), el nitrógeno orgánico disuelto (la fracción más importante está constituida por aminoácidos y péptidos, y se le denomina habitualmente N-amino), el Nitrógeno particulado orgánico (debido a pequeños organismos: algas, bacterias) y el particulado inorgánico [130]. La presencia de Nitrógeno Orgánico Disuelto (DON) en aguas naturales es en gran medida debido a procesos biológicos autóctonos. Adicionalmente, las fuentes externas de DON provienen de aguas residuales industriales, migraciones terrestres o deposición atmosférica [131]. La concentración de DON en aguas se ha ignorado durante mucho tiempo. Su medida resulta dificultosa y su carácter químico se ha descrito escasamente. Esto, unido a que se supone biológicamente inerte, pueden ser las causas [131]. Existen buenos procedimientos analíticos para cuantificar el nitrógeno inorgánico disuelto (DIN) y el nitrógeno disuelto total (TDN), pero no para el Nitrógeno orgánico disuelto, tal como se ha mencionado anteriormente. El más utilizado es el método Kjeldahl [132-135], debido a que es el de referencia establecido por la Directiva Europea [40] relativa a los métodos de medida en el análisis de aguas superficiales. El método Kjeldahl se desarrolló específicamente para el nitrógeno proteico, y algunos autores han indicado la recuperación incompleta del nitrógeno para algunos compuestos [136-137]. Este método proporciona el contenido de Nitrógeno orgánico disuelto y amonio. En otros protocolos el DON se calcula como la diferencia entre las medidas independientes del TDN y el DIN, pero estos métodos son incluso más largos en tiempo que el método de Kjeldahl. Los principales métodos para la determinación del TDN son: la oxidación alcalina con persulfato o la fotooxidación-UV a nitrato y los métodos de combustión oxidativa a NO [131,136]. Se han publicado algunos trabajos comparando varios de estos métodos [131,138- 139], pero se necesita todavía mucho más trabajo para mejorar la exactitud de los resultados de los métodos para el DON y reducir el tiempo y/o la dificultad de la técnica. En esta parte de la Tesis, se ha profundizado en el estudio del Nitrógeno Orgánico en dos direcciones: § En una primera parte, se han demostrado las ventajas del método fluorimétrico de OPA/NAC frente al Nessler para la determinación de amonio después de la digestión Kjeldahl, además de demostrar que puede estimar directamente amonio y aminas primarias. § En la segunda parte, se propone un método quimioluminiscente que puede evaluar el nitrógeno orgánico mayoritario total de forma rápida y automática.
- 163. Capítulo 2. RESULTADOS OBTENIDOS Y DISCUSIÓN 139 2.2.2.1. MÉTODO FLUORIMÉTRICO DE ROTH MODIFICADO PARA MUESTRAS DE AGUA: DETERMINACIÓN SIMULTÁNEA DE AMONIO Y GRUPOS AMINO PRIMARIO Y SU UTILIDAD EN LA DETERMINACIÓN DEL NITRÓGENO KJELDAHL TOTAL. El amonio es un micronutriente en los sistemas acuosos, y tiene un papel fundamental en el ciclo del nitrógeno de los ecosistemas acuáticos. Sin embargo, su determinación todavía es una tarea delicada, debido a su susceptibilidad a la contaminación, y a que los métodos clásicos no parecen estar bien controlados en la mayoría de los laboratorios [140]. Habitualmente, el amonio se encuentra junto a aminas alifáticas de cadena corta (principalmente metilamina), debido a que estas se hallan ampliamente distribuidas en el medioambiente. Los límites tolerables para aminas se regulan como el Nitrógeno Kjeldahl Total (que incluye amonio y Nitrógeno Orgánico), siendo los valores habituales entre 15 y 85 mg/L para aguas residuales, y 1 mg/L para agua de grifo. La cromatografía líquida combinada [10] o no con derivatización [141], se utiliza generalmente para el análisis de amonio y aminas alifáticas en medio acuoso. En la bibliografía no se encuentran métodos UV-VIS o fluorimétricos libres de errores sistemáticos que puedan utilizarse como métodos de screening de muestras para reducir costes y tiempo de análisis en el laboratorio medioambiental, o para determinaciones in situ. El método fluorimétrico de Roth [142-144] para la determinación de aminas primarias utilizando como reactivos derivatizantes un tiol y el o-ftaldialdehido (OPA), podría adaptarse a la determinación de amonio y grupos amino primario totales. Aprovechando la respuesta selectiva de amonio con el reactivo OPA-NAC a 415 nm, que se ha demostrado en el Punto 2.1.2.1 del Capítulo 2-Resultados de esta Tesis (Apéndice 4, [55]), y que nuestro grupo de investigación ha publicado recientemente la determinación de grupos amino primario en muestras de agua llevando a cabo la derivatización de las aminas en cartuchos C18 y utilizando el reactivo OPA-NAC [145,37] y 333 nm como longitud de onda de excitación, se estudió en este trabajo la determinación simultánea de amonio y metilamina, como compuesto modelo de los grupos amino primario, utilizando el reactivo OPA-NAC como agente derivatizante, excitando a dos longitudes de onda 415 nm y 333 nm, y empleando una metodología de calibración tanto bivariada como multivarida (regresión en componentes principales). El método se aplicó a patrones antes y después del tratamiento Kjeldahl, siendo un buen indicador del nitrógeno amino primario presente en la muestra o un buen método para amonio después del tratamiento Kjeldahl, y también un buen indicador de fallos en el tratamiento de digestión Kjeldahl utilizado para la conversión a amonio de los grupos amino primario. El método se aplicó al análisis de muestras de agua reales, y los resultados se compararon con los obtenidos al aplicar el método de referencia de Nessler. Más información acerca de las condiciones experimentales, se refleja en el Apéndice 10.
- 164. REACCIÓN DEL LUMINOL 140 Etapa de calibración De acuerdo con el trabajo previo del grupo de investigación [145], los patrones de MA, EA, N-PrA, IPA, BA, PeA, HA y β-FEA, proporcionaron respuestas similares entre ellos al expresar la concentración en mg/L de Nitrógeno, con porcentajes de recuperación respecto a la señal de la MA de 100, 93.5, 101.1, 93.4, 89, 100.4, 108.2 y 99 % respectivamente. La aditividad entre la señal de diferentes aminas se evaluó analizando mezclas de estas. Los porcentajes de recuperación fueron entre el 81-88 %. Con estos resultados se demostró que la respuesta de las aminas en la reacción con OPA-NAC, no dependía de la amina primaria, y se podía utilizar la MA como compuesto modelo. Para estudiar la determinación simultánea de amonio y aminas primarias (MA), se empleó el diseño experimental 52 que se muestra en la Tabla 63. Tanto el amonio como la amina primaria proporcionaban una señal fluorescente a ëexc = 333 nm / ëem = 450 nm, aunque el máximo de emisión para el amonio se situaba a 462 nm (Figura 53). Tabla 63. Código y concentración del conjunto de patrones de calibración (diseño 5 2 ). NH4 + MA (mg/L) 0 0.25 0.375 0.5 0.625 0 00 01 02 03 04 0.25 10 11 12 13 14 0.75 20 21 22 23 24 1.25 30 31 32 33 34 1.75 40 41 42 43 44 No hubo diferencias significativas a un nivel de confianza del 95% (varianza homogénea, Fcalc < Ftab, α=0.05 , y pendientes comparables, tcalc < ttab, α=0.05 ) entre las curvas de calibrado de amina obtenidas en presencia de diferentes concentraciones de amonio constante variado de 0 a 1.75 mg/L (Tabla 64). Las pendientes medias obtenidas a 450 nm para amina y amonio fueron 6532 (s= 472, n= 5) y 1038 (s= 127, n= 5), y expresando la concentración en mg/L de N, 2950 (s= 213, n= 5) y 855 (s= 104, n=5), respectivamente. Estos valores indican que el isoindol de amina es 3.5 veces más sensible que el isoindol de amonio cuando se trabaja excitando a 333 nm y midiendo la emisión a 450 nm.
- 165. Capítulo 2. RESULTADOS OBTENIDOS Y DISCUSIÓN 141 Figura 53. Espectros de emisión de fluorescencia para patrón de NH4 + de 1.75 mg/L (a λexc =333 nm y a λexc=415 nm) y patrón de MA de 0.375 mg/L (a λexc=333 nm y a λexc=415 nm). En la Figura 53, se muestran los registros obtenidos para amonio y metilamina excitando a 415 nm. Para la amina se obtuvo una señal similar al blanco (de acuerdo con el Punto 2.1.2.1 del Capítulo 2-Resultados de esta Tesis) , mientras que para el amonio se obtuvo un máximo de emisión de fluorescencia a 485 nm. Como puede verse en la Tabla 64, a la longitud de onda selectiva para amonio, λexc = 415 nm (λem= 485 nm), se obtuvo la misma curva de calibrado en presencia o ausencia de amina. Para las diferentes cantidades de amina ensayadas, las ordenadas fueron similares a la obtenida con la curva de calibrado de patrones de amonio, debiéndose su contribución al blanco de reactivos. La curva de calibrado de amonio a 485 nm, así como las ecuaciones de calibración de amonio y de amina a 450 nm, excitando a 415 y a 333 nm respectivamente, se necesitan para la estimación de ambos analitos. 0 500 1000 1500 2000 2500 3000 3500 4000 380 430 480 530 580 λλ (nm) Fluorescencia MA λexc = 333 nm NH4 + λexc = 333 nm NH4 + λexc = 415 nm MA λexc = 415 nm
- 167. Capítulo 2. RESULTADOS OBTENIDOS Y DISCUSIÓN 143 Los espectros de emisión obtenidos excitando a 415 y 333 nm para los patrones (Tabla 63), se procesaron utilizando regresión en componentes principales (PCR) para desarrollar un modelo multivariado para amonio y amina respectivamente. Para llevar a cabo la etapa de calibración, se emplearon la Selección Top Down (TDS) y la selección del mejor Sub-Conjunto (BSS) [146]. Los modelos de PCR se obtuvieron utilizando los datos (centrados) como bloque-X. El número de variables, el porcentaje de varianza explicada, el número de componentes y la media cuadrática de los errores de predicción (RMSECV) por validación cruzada, se muestran en la Tabla 65. Los valores de RMSECV se calcularon para evaluar la capacidad de predicción de los modelos. Para la metilamina no había diferencias entre las dos opciones de selección, y se obtuvo un modelo de complejidad 3. Para el amonio, la componente principal 2, no se incluyó en el modelo con selección BSS, utilizándose solamente las componentes principales 1 y 3 (complejidad 2). En un segundo paso, se llevó a cabo una selección de las variables sobre el modelo optimizado utilizando la aproximación de Eliminación de Variables No informativas (UVE) [147]. Se utilizaron solamente 32 y 29 de las variables originales para metilamina y amonio respectivamente. En ambos casos, se seleccionó un modelo final de complejidad dos. Los valores de RMSECV fueron buenos para todos los modelos, como puede verse en la Tabla 65, pero se seleccionaron los modelos de UVE- BSS. Tabla 65. Características de los modelos de Regresión en Componentes Principales obtenidos para amonio y metilamina utilizando Selección Top Down (TDS), Selección del mejor Subconjunto (BSS) o Eliminación de las Variables no informativas-Selección del mejor Subconjunto. Amonio (ëexc 415 nm) Amina (ëexc 333 nm) TDS BSS UVE-BSS TDS BSS UVE-BSS Número de variables 231 231 32 180 180 29 CPs 1,2 1,3 1,2 1,2,3 1,2,3 1,2 Varianza Explicada, bloque-X 99.999 99.999 99.999 99.999 99.999 99.999 RMSECV 0.089 0.051 0.052 0.034 0.037 0.040 Precisión y exactitud Teniendo en cuenta los resultados previos, se analizaron mezclas de patrones de amonio y metilamina. Las medidas se llevaron a cabo excitando a 415 nm y a 333 nm, y utilizando las curvas de calibrado de amonio a 485 nm y las curvas de calibrado de amonio y de metilamina a 440 nm. En la Tabla 66 se muestran las concentraciones de amonio y metilamina calculados en las diferentes mezclas ensayadas para cada uno de los modelos de calibración utilizados. Los resultados obtenidos utilizando calibración bivariada fueron satisfactorios en todos los casos, siendo los errores relativos entre 4-24%
- 168. REACCIÓN DEL LUMINOL 144 para el amonio y entre 7-8% para la metilamina. La desviaciones estándar relativas fueron entre 1-9% y 6-13% para amonio y metilamina respectivamente. Al utilizar la calibración multivariada (PCR), las exactitud fue algo mejor para el amonio siendo los porcentajes de error relativo entre 1-16%, y similar para la metilamina, con porcentajes de error relativo entre 0-7.2%. Tabla 66. Concentración de amina y amonio hallada en mezclas de patrones aplicando calibración bivariada o multivariada (PCR). NH4 + encontrado (mg/L) MA encontrada (mg/L)NH4 + adicionado (mg/L) Calibración bivariada PCR MA adicionada (mg/L) Calibración bivariada PCR 0.25 0.31 ± 0.03 0.29 ± 0.02 0.25 0.23 ± 0.03 0.26 ± 0.02 0.75 0.89 ± 0.04 0.76 ± 0.06 0.375 0.35 ± 0.02 0.40 ± 0.06 1.25 1.14 ± 0.03 1.23 ± 0.07 0.5 0.46 ± 0.06 0.50 ± 0.04 1.75 1.68 ± 0.02 1.74 ± 0.06 0.625 0.58 ± 0.04 0.58 ± 0.09 Método Kjeldahl La determinación del Nitrógeno orgánico es un parámetro importante en la calidad del agua. Este parámetro se estima según la legislación de la diferencia entre el Nitrógeno Kjeldahl Total y el Amonio libre. Se ensayaron diferentes condiciones de digestión-destilación Kjeldahl para seleccionar aquellas que garantizaran la conversión total de las aminas (Tabla 67). Las variables ensayadas fueron: cantidad de HgO (g) (0.0332-0.525 g), volumen de H2SO4 concentrado (3.39-12 mL), cantidad de K2SO4 (2.22-10.5g), tiempo de digestión (90-120 minutos) y volumen del reactivo utilizado en el paso de destilación NaOH/Na2S2O3 (25- 50 mL). Se utilizan los modelos previamente establecidos para la determinación de amonio y amina no transformada. Mientras que el amonio extraído fue prácticamente independiente de las condiciones experimentales (Figura 54), la trasformación de amina en amonio, varió en función de las condiciones de digestión-destilación empleadas, como puede verse en la Figura 55. La condición 6 proporcionó la transformación total de MA a amonio, siendo el porcentaje de conversión del 109%. El porcentaje de conversión obtenido con el método de Nessler en la condición 6, también fue del orden del 100%.
- 169. Capítulo 2. RESULTADOS OBTENIDOS Y DISCUSIÓN 145 Figura 54. %Recuperación de NH4 + calculado respecto a la señal(o pendiente) obtenida sin Kjeldahl, a diferentes condiciones Kjeldahl (Tabla 67). Condición 2: n=7 : 5 patrones de 0.5 mg/L de NH4 + y 2curvas de calibrado (0, 0.5, 1.25, 2, 2.75 mg/L NH4 + y 0, 0.2, 0.5, 0.9, 1.3 mg/L NH4 + respectivamente). Condición 3: n=4: 2 patrones de 0.5 mg/L de NH4 + y 2 curvas de calibrado (0, 0.05, 0.25, 0.78 mg/L NH4 + , y 0, 0.75, 0.75 mg/L NH4 + ). Condición 5: n=1: Curva de calibrado (0, 0.28, 0.56 mg/L NH4 + ). Condición 6: n=3: Patrones de 0.375 y 0.75 (2 réplicas) mg/L NH4 + . Figura 55. %Recuperación de MA transformada a amonio y %Recuperación de MA no transformada, calculados respecto a la señal obtenida sin Kjeldahl, a diferentes condiciones Kjeldahl (Tabla 67). Condición 1: n=4 : 2 patrones de 0.5 mg/L y 2 patrones de 0.125 mg/L de MA. Condición 2: n=4: Patrones de MA de 0.086, 0.3875, 0.5 y 0.56 mg/L. Condición 4: n=2: Patrón de 0.5 mg/L de MA (2 réplicas). Condición 5: n=5: Patrón de 0.775 mg/L de MA (5 réplicas). Condición 6: n=2: Patrón de 1.3 mg/L de MA (2 réplicas). 0 20 40 60 80 100 120 140 160 2 3 5 6 Condición %Recuperación 440 nm 485 nm 0 20 40 60 80 100 120 140 1 2 4 5 6 Condición %Recuperación MA 440 nm NH 485 nm
- 170. REACCIÓN DEL LUMINOL 146 Tabla 67. Diferentes condiciones Kjeldahl ensayadas. Condición m(K2SO4) (g) m(HgO) (g) V(H2SO4) (mL) Tiempo de Digestión (min) V(NaOH/Na2S2O3) (mL) 1 2.224 0.0332 3.39 90 25 2 2.224 0.0332 3.39 120 25 3 10.5 0.525 12 120 50 4 6.7 0.1 10.2 120 30 5 6.7 0.1 10 120 40 6 6.7 0.525 10 120 30 Además se estudió la conversión de otras aminas primarias y secundarias (etilamina, n-propilamina, isopropilamina, butilamina, pentilamina, hexilamina, dimetilamina, dietilamina y β-feniletilamina) a amonio en las condiciones óptimas de digestión-destilación Kjeldahl (Condición 6). En la Figura 56 se muestran los espectros de emisión de fluorescencia a ëexc = 415 nm (selectiva para amonio) de los derivados OPA-NAC-amonio y OPA-NAC-amina a diferentes niveles de concentración antes del tratamiento Kjeldahl, y a concentración de amonio de 0.75 mg/L después del tratamiento Kjeldahl y la derivatización con OPA-NAC. Como puede verse en la Figura 56-A, la señal analítica de los derivados de amina antes de la digestión Kjeldahl, fue similar a la del blanco. Sin embargo, después del tratamiento Kjeldahl, las aminas se transformaron en amonio (Figura 56-B). Las medidas del amonio se llevaron a cabo por el procedimiento OPA-NAC (λexc 415 nm - λem 485 nm) y se contrastaron con el método de Nessler. Los porcentajes de recuperación de amonio obtenidos con ambos métodos se muestran en la Tabla 68. Como puede observarse, con ambos métodos fueron comparables, y los porcentajes de recuperación fueron cercanos al 100%, tanto en disoluciones de una única amina como en mezclas. Tabla 68. Porcentajes de recuperación de amonio para las diferentes aminas y mezclas de aminas después del Kjeldahl, con el Método OPA-NAC a 485 nm o con el Método Nessler. Amina (Concentración antes Kjeldahl) Método OPA-NAC (%RECNH4+ ) Método Nessler (%RECNH4+ ) EA (3.75 mg/L) 101 110 N-PrA (4.93 mg/L) 122.5 121.5 IPA (4.93 mg/L) 107.2 103.13 BA (6.10 mg/L) 88.2 81.8 PeA (7.25 mg/L) 99.8 96 HA (8.43 mg/L) 124.7 121 DMA(3.76mg/L) 71.5 68.6 DEA(6.10 mg/L) 112 101.2 β-FEA(10.10 mg/L) 104.4 100.7 EA (1.878 mg/L) + DEA (3.047 mg/L) 84.4 79 DMA (1.253 mg/L) + N-PrA (1.642 mg/L) + IPA (1.642 mg/L) 82 81 BA (3.05 mg/L) + β−FEA (5.05 mg/L) 99.9 85 PeA (3.622 mg/L) +HA (4.21 mg/L) 85.3 92
- 171. Capítulo 2. RESULTADOS OBTENIDOS Y DISCUSIÓN 147 Figura 56. Espectro de emisión de fluorescencia de los derivados OPA-NAC a 485 nm (ëexc = 415 nm) para blanco, patrón de 0.75 mg/L de NH4 + , 1.3 mg/L MA, 1.875 mg/L EA y DMA, 2.465 mg/L N-PrA e IPA, 3.05 mg/L BA y DEA, 3.625 mg/L PeA, 4.215 mg/L HA, y 5.05 mg/L β-FEA. (56-A)-medidos antes del Kjeldahl. (56-B) medidos después del Kjeldahl. 450 500 550 600 0 200 400 600 800 1000 DMA BA HA, NPrA Patrón 0.75 mg/l NH4+, MA, EA, IPA, PeA, DEA, B-FEA B) Fluorescencia λem (nm) Blanco 450 500 550 600 0 200 400 600 800 1000 FluorescenceSignal λem (nm) Patrón 0.75 mg/l NH4+ Blanco, MA, EA, NPrA, IPA, BA, PeA, HA, DMA, DEA, B-FEA A)
- 172. REACCIÓN DEL LUMINOL 148 Análisis de muestras de agua El procedimiento se aplicó a la determinación de amonio y aminas primarias (expresadas como metilamina) en tres muestras de agua reales de diferente naturaleza: agua de riego, residual y de fuente. En la Tabla 69 se muestran las concentraciones de amonio y amina halladas aplicando cada uno de los modelos de calibración. Para comparar los resultados obtenidos, se aplicó un test t de muestras relacionadas, que dio á > 0.05, tanto para el amonio como para la metilamina, lo que indicaba que los resultados eran similares en ambos métodos, y que se podía optar por cualquiera de los dos modelos de calibración, para el análisis de muestras reales. La muestra de agua de fuente se analizó además por el método de referencia de Nessler obteniéndose resultados comparables para la concentración de amonio: (0.32 ± 0.02) mg/L (n=3). Tabla 69. Concentración de amina y amonio hallada en muestras reales aplicando calibración bivariada o multivariada (PCR). NH4 + (mg/L) Amina (expresado como MA) (mg/L) Muestras Calibración Bivariada PCR Calibración Bivariada PCR Agua de riego - - 0.051 ± 0.007 0.086 ± 0.009 Agua residual 24.92 ± 0.10 32.2 ± 0.4 8.8 ± 0.4 10.25 ± 0.14 Agua de fuente 0.36 ± 0.03 0.29 ± 0.05 0.058 ± 0.013 0.082 ± 0.004 El procedimiento OPA-NAC se aplicó a estas tres muestras de agua después del tratamiento de digestión Kjeldahl. La señal analítica medida correspondió al contenido de amonio más el nitrógeno orgánico. Los resultados obtenidos se compararon con los que proporcionó el método de Nessler. Al aplicar el método OPA-NAC los resultados fueron (0.19 ± 0.07, n=3), (213 ± 3, n=3) y (2.103 ± 0.018, n=3) mg/L de amonio para las muestras de agua de riego, residual y de fuente respectivamente. Estos resultados fueron estadísticamente similares (test t de muestras relacionadas, á > 0.05) a los obtenidos al aplicar el método de referencia de Nessler: (0.223 ± 0.018, n=3), (232 ± 3, n=3) y (2.14 ± 0.02, n=3) mg/L de amonio. Las concentraciones de amonio halladas en las muestras, están de acuerdo con la diferente naturaleza de las aguas analizadas. Para aguas superficiales de baja contaminación el N-Kjeldahl no debe exceder las 2 mg/L. Para aguas residuales industriales, este parámetro puede ser incluso superior a 200 mg/L. Teniendo en cuenta los resultados mostrados en la Tabla 69 (concentraciones antes del Kjeldahl) y los resultados después del tratamiento Kjeldahl, se dedujo que estas muestras de agua, presentaban otros compuestos de nitrógeno además de amonio y aminas alifáticas primarias.
- 173. Capítulo 2. RESULTADOS OBTENIDOS Y DISCUSIÓN 149 Conclusiones En este capítulo, se propone la utilización del reactivo OPA-NAC para la determinación simultánea de amonio y aminas alifáticas primarias. Puede utilizarse calibración bivariada a dos longitudes de onda de excitación, o calibración multivariada (PCR), obteniéndose resultados similares. La metilamina se utilizó como compuesto modelo debido a que la respuesta de diferentes aminas fue la misma cuando se expresaba en mg/L de Nitrógeno, y se demostró la aditividad entre sus señales cuando se procesaron mezclas de aminas. Las curvas de calibrado de amonio en el intervalo 0.25-1.75 mg/L en presencia de una concentración fija de amina (variada de 0.25 a 0.625 mg/L) dieron pendientes estadísticamente similares excitando a 415 nm. A esta longitud de onda, no había respuesta por parte de las aminas. Las curvas de calibrado de metilamina en el intervalo 0.25-0.625 mg/L en presencia de una cantidad fija de amonio (variada de 0.25 a 1.75 mg/L), también dieron pendientes estadísticamente similares excitando a 333 nm. La señal del amonio afectaba a los valores de la ordenada. Las pendientes de las curvas de calibrado de amonio fueron similares antes y después de aplicar el tratamiento Kjeldahl. La conversión de aminas a amonio fue aproximadamente del 100% en las condiciones óptimas establecidas para todas las aminas individualmente y en mezclas. La aplicación del tratamiento Kjeldahl a las muestras de agua reales, dio resultados de acuerdo con la naturaleza de las aguas. La aplicación del método de referencia de Nessler sirvió para validar el método, obteniéndose resultados similares a los obtenidos con el método OPA-NAC.
- 174. REACCIÓN DEL LUMINOL 150 2.2.2.2. MÉTODO QUIMIOLUMINISCENTE AUTOMÁTICO PARA LA ESTIMACIÓN TOTAL DEL NITRÓGENO ORGÁNICO (-CH2-NH-) Y AMONIO. Basándonos en la bibliografía reciente para amonio [148] y en la conocida reactividad entre los grupos amino y el hipoclorito, en este capítulo se propone un sistema de flujo continuo para la determinación conjunta del nitrógeno orgánico amino (-CH2- NH-) y el nitrógeno amoniacal en muestras de agua no tratadas. En este sistema, el grupo amino reacciona con el Hipoclorito formando la cloramina, y posteriormente, la mezcla reacciona con el Luminol generando una señal quimioluminiscente. La señal de emisión, registrada a 425 nm en función del tiempo, disminuye al aumentar la concentración de Nitrógeno en la muestra, debido a la disminución en la concentración de Hipoclorito, que depende de la concentración de grupos amino presentes en la muestra. Se han ensayado un gran número de compuestos de Nitrógeno, y sus sensibilidades se han comparado en (mg/L)-1 de Nitrógeno. Una misma curva de calibrado puede utilizarse para un gran número de compuestos, con lo que el método puede emplearse para deducir de forma rápida el Nitrógeno orgánico disuelto y el amonio. Se han evaluado las características del método como la selectividad, tiempo de análisis y reproducibilidad. El método quimioluminiscente se ha aplicado al análisis de una amplia variedad de muestras de agua (agua natural, de lago, de riego, de fuente, residual y marina). La exactitud del método se ha evaluado aplicando el método de adición estándar y procedimientos de referencia. Para algunas de estas muestras, se ha realizado un estudio comparativo del contenido de Nitrógeno amoniacal antes y después del tratamiento Kjeldahl, para evaluar si el método aplicado a muestras no tratadas podía dar una estimación del N Kjeldahl Total. El sistema de inyección en flujo utilizado es el que se muestra en la Figura 18 del Capítulo 1-Introducción (Pág. 41). Más información acerca de las condiciones experimentales, se refleja en el Apéndice 11. Parámetros analíticos En la Tabla 70, se muestran las curvas de calibrado Señal QL frente a Concentración de Nitrógeno (mg/L) obtenidas con patrones de amonio en diferentes condiciones experimentales. Las variables evaluadas fueron: el tipo de celda de medida (celda de nudos o espiral), la concentración de tampón, la concentración de hipoclorito, y la presencia de tampón o de NaCl en los patrones. Las condiciones óptimas, de máxima sensibilidad (marcadas en negrita en la Tabla 70), se obtuvieron utilizando la celda de nudos e Hipoclorito 0.9 mM en tampón carbonato 0.144 mM de pH 11.2. El rango lineal fue de 0.23-4 mg/L N, y el límite de detección alcanzado fue de 0.070 mg/L N. Al acondicionar los patrones con tampón, la sensibilidad disminuyó un 33%, y al prepararlos en NaCl, disminuyó un 13%.
- 175. Capítulo 2. RESULTADOS OBTENIDOS Y DISCUSIÓN 151 Tabla 70. Curvas de calibrado de amonio (Señal QL frente a Concentración de Nitrógeno (mg/L)) y otros parámetros analíticos en diferentes condiciones experimentales estudiadas. Condiciones estudiadas Celda de detección Conc. Hipoclorito Conc. Tampón Curva de calibrado Y = (a ±± sa) + (b ±± sb)· C (n, r2 , sy/x) LD (mg/l) IL (mg/l) Espiral 0.9 mM 0.144 mM Y = (9170 ± 30) + (-1844 ± 14)· C (12, 0.9994, 70) 0.091 0.3-4 Nudos 0.9 mM 0.144 mM Y = (9680 ±± 140) +(-2370 ±± 70)· C (10, 0.9938, 300) 0.070 0.24-4 Nudos 0.5 mM 0.144 mM Y = (4300 ± 50) + (-1960 ± 40)· C (9, 0.9968, 100) 0.086 0.3-2 Nudos 0.9 mM 0 mM Y = (9690 ± 100) +(-2290 ± 40)· C (12, 0.9962, 200) 0.073 0.24-4 Nudos 0.9 mM 10 mM Y = (9400 ± 90) + (-1730 ± 40)· C (12, 0.9940, 200) 0.097 0.3-4 Nudos* 0.9 mM 0.144 mM Y = (7960 ± 160) + (-1570 ±70)· C (12, 0.9806, 400) 0.107 0.4-4 Nudos** 0.9 mM 0.144 mM Y = (8290 ± 100) + (-2050 ±40)· C (15, 0.9942, 200) 0.082 0.27-4 * Tampón en patrones **NaCl en patrones Los registros de inyección en flujo obtenidos para los patrones del calibrado de amonio, se muestran en la Figura 57. Figura 57. Registros de inyección en flujo obtenidos en las condiciones experimentales óptimas a diferentes niveles de concentración de amonio (expresado en mg/L de Nitrógeno). 0 100 200 300 400 0 2000 4000 6000 8000 10000 SeñalQuimioluminiscente Tiempo (s) 0.75 mg/l N Blanco 2 mg/l N 3 mg/l N 4 mg/l N
- 176. REACCIÓN DEL LUMINOL 152 La reproducibilidad del método se evaluó en dos vertientes. En la Figura 58 se muestra el solapamiento a todos los niveles de concentración entre las curvas de calibrado de amonio obtenidas en cinco días diferentes. El porcentaje de desviación estándar relativa (%DER) para la pendiente del calibrado fue de 1.86 %. Figura 58. Curvas de calibrado de amonio (Señal QL frente a mg/L N) obtenidas en cinco días diferentes. Por otra parte, los estudios de repetibilidad dieron %DER(n=3) de 0.81%, 0.31% y 3.7% para concentraciones de Nitrógeno de 0.75, 2 y 4 mg/L respectivamente. Estos valores indicaron que la precisión del método era muy buena. La velocidad de muestreo de hasta 60 muestras por hora sin tratamiento previo, permite que el procedimiento pueda adaptarse como analizador para monitorización in situ. Respuesta de compuestos de Nitrógeno Para evaluar la validez de la metodología propuesta para la determinación de los grupos amino (Nitrógeno Orgánico más Nitrógeno amoniacal), se estudió la respuesta de otras familias de compuestos de Nitrógeno, comparando los resultados con los obtenidos para el amonio, refiriendo las concentraciones a contenido en Nitrógeno. La primera familia de compuestos estudiada fueron las aminas alifáticas: primarias, secundarias y terciarias. y = -2293.3x + 9365.4 y = -2255.9x + 9570.4 y = -2332.4x + 9456 y = -2339.4x + 9677.6 y = -2366.3x + 9676 0 2000 4000 6000 8000 10000 12000 0 1 2 3 4 5 Concentración de Nitrógeno (mg/L) SeñalQuimioluminiscente Día 1 Día 2 Día 3 Día 4 Día 5 Curvas de calibrado de Amonio
- 177. Capítulo 2. RESULTADOS OBTENIDOS Y DISCUSIÓN 153 El estudio de siete aminas alifáticas primarias (MA, EA, N-PrA, IPA, BA, PeA y HA), dio porcentajes de recuperación (calculados como pendiente de la curva de calibrado de la amina*100/pendiente de la curva de calibrado de amonio) cercanos al 100% como puede verse en la Tabla 71. El porcentaje de recuperación medio (91 ± 11)% demostró que la respuesta de las aminas primarias era similar a la del amonio, por lo que su contenido de nitrógeno quedaría medido utilizando esta metodología. Para las aminas alifáticas secundarias, DMA y DEA, los porcentajes de recuperación fueron algo menores (Tabla 71) aunque cercanos al 100%, siendo el porcentaje de recuperación medio (81 ± 4) %. Tabla 71. Porcentajes de recuperación (bamina*100/bNH4+) para aminas alifáticas primarias y secundarias individualmente y %Recuperación medio. Amina %Recuperación bamina*100/bNH4+ (%REC ± s) MA 104 EA 83 N-PrA 79 IPA 82 BA 82 PeA 102 HA 103 (91 ± 11)% DMA 78 DEA 84 (81 ± 4)% Sin embargo, los resultados para las dos aminas alifáticas terciarias estudiadas (TEA y TMA), mostraron que este tipo de compuestos no se podían detectar mediante esta metodología. Esto se debe a que en estas condiciones experimentales, la cloramina no se formaba y por tanto el hipoclorito no se consumía, permaneciendo la señal quimioluminiscente constante, igual al blanco. La segunda familia de compuestos estudiados fueron las poliaminas. Estos compuestos presentan varios grupos amino y la respuesta para cada analito debería disminuir en función del número de grupos amino presentes. Al realizar la calibración frente al contenido de nitrógeno para cada una de las poliaminas estudiadas (putrescina, cadaverina, espermina, espermidina y 1,7-diaminoheptano), se obtuvieron porcentajes de recuperación excelentes (Tabla 72). El porcentaje de recuperación medio (96.6 ± 1.8) %, indicó que el contenido de nitrógeno de este tipo de compuestos quedaría medido utilizando esta metodología. La tercera familia de compuestos de nitrógeno estudiada fueron las arilaquilaminas. Se tomó la β-Feniletilamina como compuesto modelo, obteniéndose también un porcentaje de recuperación del 100.1%.
- 178. REACCIÓN DEL LUMINOL 154 Tabla 72. Porcentajes de recuperación (bpoliamina*100/bNH4+) para poliaminas individualmente y %Recuperación medio. Poliamina % Recuperación bpoliamina*100/bNH4+ (% REC ± s) Putrescina 98 Cadaverina 95 Espermina 98 Espermidina 94 1,7-Diaminoheptano 97 (96.6 ± 1.8)% Por otra parte, se estudiaron compuestos de Nitrógeno que contenían Nitrógenos aromáticos (como la triazina) o Nitrógenos resonantes (como la urea). Estos Nitrógenos no respondían a la reacción con el Hipoclorito para dar cloramina, de modo que la señal quimioluminiscente permanecía estable (similar al blanco). Además los Nitrógenos aromáticos, exaltaban la señal quimioluminiscente del blanco. Estos resultados mostraron que Nitrógenos resonantes y aromáticos no podrían ser detectados mediante este método. Además se estudiaron 14 aminoácidos que solo poseían un grupo amino. Los porcentajes de recuperación obtenidos (Tabla 73), fueron cercanos al 100% en todos los casos, siendo el porcentaje de recuperación medio de (96 ± 4) %. Tabla 73. Porcentajes de recuperación (baminoacido*100/bNH4+) para aminoácidos individualmente y %Recuperación medio. Aminoácido % Recuperación baminoácido*100/bNH4+ (% REC ± s) Ácido aspártico 93.5 Triptófano 87.1 Ácido glutámico 101.9 Glicocola 97.6 Prolina 96.6 Serina 99.9 Tirosina 95.4 Almandina 98.1 Fenilalanina 97.1 Valina 100.6 Leucina 94.3 Isoleucina 96.0 Treonina 97.1 Lisina 93.8 (96 ± 4)%
- 179. Capítulo 2. RESULTADOS OBTENIDOS Y DISCUSIÓN 155 El aminoácido Histidina presenta una estructura con dos nitrógenos aromáticos además del grupo amino alifático, y al igual que para la triazina, la señal quimioluminiscente del blanco aumentó y no se observó disminución de señal en el intervalo de trabajo del Nitrógeno. Por su parte, el aminoácido Arginina que presenta Nitrógenos resonantes además del grupo amino alifático, los resultados obtenidos considerando solamente el nitrógeno alifático (baminoácido*100/bamonio=96.8%), confirmaron que solo este grupo (–CH2-NH2) de la arginina contribuyó a la disminución de la señal quimioluminiscente. Los aminoácidos Cisteína y Metionina dieron porcentajes de recuperación (baminoácido*100/bamonio) de 287 y 147 %. Estos porcentajes indicaron que otro átomo de su estructura contribuía a disminuir la señal quimioluminiscente. Como se demuestra más abajo, este átomo fue el S. Selectividad del método: Estudio de interferencias El tipo de compuestos interferentes estudiados fueron aquellos que contenían átomos de sulfuro, ya que estos átomos podían dar lugar a interferencias en la determinación de nitrógeno como se había visto en la sección anterior al estudiar los aminoácidos que contenían azufre en su estructura. Se estudió la respuesta de dos compuestos, el sulfuro sódico (S2- ) y del ácido mercaptoacético (R-SH) que no contenían grupos nitrógeno en su estructura. Ambos compuestos respondieron a la reacción disminuyendo la señal QL. La interferencia del sulfuro fue el doble que la del mercaptoacético, siendo las pendientes de las curvas de calibrado 3058 y 1850 L/mg S. La respuesta de aminoácidos como la Cisteína (aminoácido con grupo R-SH en su estructura) y su derivado la N-Acetil-L-Cisteína, era debida a la contribución de ambos átomos, N y S. Las señales experimentales fueron próximas al 100% de las señales teóricas obtenidas teniendo en cuenta las pendientes de calibración de amonio y mercaptoacético. La interferencia causada por el grupo R-S-R, es pequeña, como puede deducirse del porcentaje de recuperación dado en la sección anterior para la metionina. Cuando se trabajó a la concentración habitual de tioles en aguas (0.25 µg/L) [149], y a la concentración máxima permitida para H2S en aguas de bebida (50 µg/L) [40], los compuestos de azufre no interfirieron en la reacción, obteniéndose una señal quimioluminiscente igual a la del blanco. El segundo grupo de posibles interferentes ensayado fueron los iones metálicos. Cr(III) y Co(II) no interfirieron en la reacción incluso a niveles de 0.5 mg/L. Cu(II) solamente disminuyó la señal QL en un 10% cuando se utilizó a una concentración de 50 µg/L (límite máximo permitido para aguas naturales). Otros cationes como Fe(III), Cd(II) y Hg(II), no interfirieron a su máxima concentración permitida (0.5 mg/L, 5 µg/L y 1µg/L respectivamente).
- 180. REACCIÓN DEL LUMINOL 156 Tratamiento Kjeldahl Se aplicó el tratamiento Kjeldahl a los compuestos de Nitrógeno que no respondían a la reacción con el Hipoclorito, para estudiar su reactividad después de la digestión de la muestra. Se digirieron patrones de trietilamina, urea, Histidina y Arginina con una concentración de Nitrógeno final de 1.5 mg/L. Los porcentajes de recuperación obtenidos se muestran en la Tabla 74. Todos los porcentajes son cercanos al 100%, por lo que después del tratamiento Kjeldahl, el contenido de Nitrógeno de estos compuestos quedaría medido al utilizar este método de detección. Además, se aplicó el tratamiento Kjeldahl a dos mezclas de aminas (que contenían o no TMA) para estudiar la recuperación del Nitrógeno Total. Los porcentajes de recuperación obtenidos para las dos mezclas con un contenido de nitrógeno total de 1.5 y 1.8 mg/L N, se muestran en la Tabla 74, siendo ambos valores cercanos al 100%. Tabla 74. %Recuperación individual y %Recuperación medio hallado en patrones de TEA, urea, Histidina y arginina de concentración final de Nitrógeno de 1.5 mg/L, y para mezclas de MA/PeA/DMA (Conc final Nitrógeno 1.5 mg/L) y MA/PeA/DMA/TMA (Conc final Nitrógeno 1.8 mg/L), después del tratamiento Kjeldahl. Analito % Rec (% REC ± s) TEA 94.1 Urea 103.5 Histidina 114.9 Arginina 109.6 MA + PeA + DMA 96.3 MA + PeA + DMA + TMA 99.7 (103 ± 8)% El tratamiento Kjeldahl se aplicó también a una proteína, la albúmina bovina. Los resultados antes y después del Kjeldahl fueron diferentes, de modo que casi la mitad del nitrógeno presente, no se detecta si no se lleva a cabo la digestión ácida. Análisis de muestras de agua Se analizaron varias muestras de agua. Para validar la exactitud del método, se fortificaron todas las muestras a tres niveles de concentración diferentes para aplicar el método de adición estándar, siendo los porcentajes de recuperación del nitrógeno adicionado muy cercanos al 100% (Tabla 75). Esta tabla demuestra que no había efecto matriz en estas muestras, y que el contenido de Nitrógeno en las muestras podía deducirse directamente a partir de la curva de calibrado de patrones. En tres de las muestras ensayadas, agua natural (S1), agua de grifo (S2) y agua de riego 2 (S3), la concentración de Nitrógeno estuvo por debajo del límite de detección del método. El agua de mar (S5) presentó un aumento de la señal del blanco por lo que no pudo ser analizada por el método por medida directa. En las otras cuatro muestras, agua
- 181. Capítulo 2. RESULTADOS OBTENIDOS Y DISCUSIÓN 157 de lago (S4), agua residual (S6), agua de riego 1 (S7) y agua de fuente (S8), se encontró una concentración de Nitrógeno detectable al realizar la medida directa de las muestras. En la Tabla 76, se muestra la concentración de Nitrógeno Total hallada en la medida directa de estas muestras calculada a partir de la curva de calibrado de patrones. Tabla 75. Concentración de Nitrógeno hallado en muestras de agua reales fortificadas y porcentajes de recuperación a diferentes niveles de concentración. Muestra Concentración adicionada (mg/L N) Concentración hallada (mg/L N) % Recuperación (Rec ± s) % 0.75 0.69 ± 0.04 92 ± 6 1.5 1.52 ± 0.05 101 ± 4S1-Agua natural 3 2.91 ± 0.08 97 ± 3 0.75 0.64 ± 0.07 86 ± 10 1.5 1.46 ± 0.02 97.4 ± 1.3S2-Agua de grifo 3 2.75 ± 0.08 92 ± 3 0.75 0.71 ± 0.03 95 ± 4 1.5 1.58 ± 0.03 105.2 ± 1.7S3-Agua de riego2 3 3.004 ± 0.018 100.1 ± 0.6 0.75 0.724 ± 0.018 97 ± 2 1.5 1.69 ± 0.04 112 ± 3S4-Agua de lago 3 2.62 ± 0.04 87.4 ± 1.3 0.75 0.76 ± 0.04 101 ± 6 1.5 1.58 ± 0.05 105 ± 3S5-Agua de mar 3 2.85 ± 0.03 95.0 ± 1.1 0.75 0.692 ± 0.017 92 ± 2 1.5 1.43 ± 0.08 96 ± 6S6-Agua residual 3 2.696 ± 0.004 89.86 ± 0.12 0.75 0.79 ± 0.03 105 ± 4 1.5 1.581 ± 0.013 105.4 ± 0.9S7-Agua de riego1 3 3.034 ± 0.007 101.1 ± 0.2 1.5 1.49 ± 0.10 99 ± 7 S8-Agua de fuente 3 2.91 ± 0.10 97 ± 3 Tabla 76. Concentración de Nitrógeno Total (Orgánico más amoniacal) (mg/L N) encontrada en las diferentes muestras de agua estudiadas, antes y después del tratamiento Kjeldahl. Concentración de Nitrógeno Total (mg/L) Muestra Antes del Kjeldahl Después del Kjeldahl S4 (0.92 ± 0.15) (1.59 ± 0.06) S5 N.D. (1.66 ± 0.10) S6 (140 ± 8) (149 ± 11) S7 (0.52 ± 0.04) (0.57 ± 0.03) S8 (0.35 ± 0.05) (1.99 ± 0.07) N.D.: No detectable.
- 182. REACCIÓN DEL LUMINOL 158 Además, se determinó el contenido de Nitrógeno de algunas de estas muestras de agua después de aplicar el tratamiento Kjeldahl. Los resultados de la Tabla 76 muestran la presencia de dos grupos de muestras: - Por una parte, las muestras cuyo contenido de Nitrógeno es el mismo antes y después del tratamiento Kjeldahl (como es el caso de las muestras S6 y S7). La muestra S6 se analizó además por los métodos de Nessler y o-ftaldialdehido/N-Acetil-L-Cisteína (OPA/NAC) para amonio que se desarrolla en el Punto 2.1.2.1 del Capítulo 2- Resultados de esta Tesis (Apéndice 4, [55]), y después del tratamiento Kjeldahl, las concentraciones halladas fueron: (180 ± 3, n=3) y (166 ± 3, n=3) mg/L N para los métodos de Nessler y OPA/NAC respectivamente. En este tipo de muestras, el tratamiento de Kjeldahl podría haberse sustituido por la aplicación sencilla y directa delsistema FIA presentado en este trabajo. - Por otra parte, las muestras que presentaban concentración de Nitrógeno diferente antes y después del Kjeldahl (como es el caso de las muestras S4, S5 y S8). La muestra S4 presentó de forma directa el 60% de la señal del N-Kjeldahl; la muestra S5 no podía analizarse de forma directa porque presentaba un aumento de señal respecto al blanco como se ha expuesto previamente; y en cuanto a los resultados para la muestra S8, las diferencias podrían explicarse debido a la presencia de compuestos de Nitrógeno que no reaccionaban con el hipoclorito pero que se transformaban en amonio después del tratamiento Kjeldahl, o más probablemente debido a la presencia de matrices orgánicas complejas que se descomponían en la digestión ácida (de forma similar a los resultados de la albúmina). Esta muestra, S8, se analizó además por los métodos de Nessler y OPA-NAC para amonio (Apéndice 4, [55]), siendo los resultados (0.25±0.02, n=3) y (0.29±0.02, n=3) mg/L N, respectivamente antes del tratamiento Kjeldahl, y (1.66 ± 0.02, n=3) y (1.635 ± 0.18, n=3) mg/L N, respectivamente después del tratamiento Kjeldahl. Estos resultados son bastante similares a los obtenidos con el procedimiento propuesto (Tabla 76). Conclusiones La respuesta de los compuestos de Nitrógeno tipo amino (-CH2-NH-) incluido el amonio, al aplicar la metodología propuesta dio porcentajes de recuperación cercanos al 100% para aminas alifáticas primarias y secundarias, poliaminas, arilaquilaminas, y para 14 de los aminoácidos ensayados. Las aminas terciarias y los Nitrógenos resonantes o los aromáticos no dieron la reacción, y las proteínas nitrogenadas dieron recuperaciones incompletas. Los principales interferentes de la reacción fueron los grupos tiol, pero se demostró que no interferían a la concentración máxima permitida por la legislación. De los iones metálicos estudiados, ninguno de ellos presentó interferencia a los máximos niveles permitidos. El método permite hacer una estimación del N-amino Total disuelto en la muestras de forma muy rápida.
- 183. Capítulo 2. RESULTADOS OBTENIDOS Y DISCUSIÓN 159 Al fortificar las muestras de agua reales, no se observó efecto matriz y los resultados obtenidos fueron exactos, con recuperaciones cercanas al 100%. Los resultados mostrados permiten establecer que para algunos tipos de muestras el tratamiento Kjeldahl se puede sustituir por el sistema de inyección en flujo presentado en este trabajo. Esta opción es muy ventajosa dado que el método no requiere tratamiento de la muestra, y se pueden procesar tres réplicas de la muestra en 1 minuto, la toxicidad de los reactivos es baja y el método se puede utilizar para monitorización in situ. La metodología propuesta también puede utilizarse como alternativa para la detección final del amonio después del tratamiento de digestión Kjeldahl de las muestras. La precisión del método para la determinación de amonio fue muy buena, con valores de %DER del 0.3-3.7% para la repetibilidad, y del 1.9% para la reproducibilidad de las pendientes de las curvas de calibración. Estos valores fueron similares a los valores de precisión obtenidos mediante la aplicación del método de Nessler (%DER=0.4%), y mejores que los obtenidos aplicando el método de OPA-NAC (%DER<12.5%). El intervalo dinámico lineal del método fue de 0.23-4 mg/L N. Para el método de Nessler, fue de 0.66-3.88 mg/L N, y para el método de OPA-NAC fue de 0.155-1.088 mg/L N. El límite de detección alcanzado con el método propuesto (0.07 mg/L N) fue similar al obtenido con el método de OPA-NAC (0.054 mg/L N), y mejor que el alcanzado con el método de referencia de Nessler (0.46 mg/L N). La rapidez del método resultó excelente (60 muestras/hora) comparada con los métodos de OPA/NAC (10 muestra/hora) o Nessler (6 muestras/hora).
- 185. Capítulo 2. RESULTADOS OBTENIDOS Y DISCUSIÓN 161 2.3. REFERENCIAS BIBLIOGRÁFICAS [1] L.Pan, J.Michael Chong, J.Pawliszyn, J.Chromatogr.A, 773 (1997) 249. [2] M.Abalos, J.M.Bayona, F.Ventura, Anal.Chem., 71 (16) (1999) 3531. [3] C.Maris, A.Laplanche, J.Morvan, M.Bloquel, Water.Sci.Technol., 40(6) (1999) 141. [4] C.Maris, A.Laplanche, J.Morvan, M.Bloquel, J.Chromatogr.A., 846 (1-2) (1999) 331. [5] F.Sacher, S.Lenz, H.J. Brauch, J.Chromatogr. A., 764 (1) (1997) 85. [6] H.Wang, J.Lin, X.Liu, H.S.Zhang, Anal.Chim.Acta, 423 (2000) 77. [7] J.Verdu Andrés, P.Campíns Falcó, R.Herraez Hernandez, Analyst, 126 (2001) 1683. [8] J.M.You, W.J.Lao, G.J.Wang, Analyst, 124 (12) (1999) 1755. [9] M. Cobo, M.Silva, J.Chromatogr. A, 848 (1999) 105. [10] D.Sahasrabuddhey, A.Jain, K.K.Verma, Analyst, 124 (1999) 1017. [11] M.Yamaguchi, S.Hara, J.Aoki, K.Yoshikuni, T.Iwata, Anal.Sci., 14(2) (1998) 425. [12] D.Djozan, M.A. Faraj-Zadeh, J. High. Resolut. Chromatogr., 19 (11) (1996) 633. [13] J.Verdu Andrés, P.Campíns Falcó, R.Herraez Hernandez, Chromatographia, 55 (2002) 129. [14] W.H.Matchett, W.C.Brumley, J. Liq. Chromatogr.Rel.Technol., 20 (1997) 79. [15] N.Seiler, Handbook of derivatives for Chromatography, Jonh Wiley, New York, 1998. [16] J.M Roselfeld., J. Chromatogr. A, 843 (1999) 19. [17] C.Molins-Legua, P.Campins Falcó, A.Sevillano Cabeza, M. Pedrón Pons, Analyst, 124 (1999) 477. [18] C.Molins Legua, P.Campíns Falcó, A.Sevillano Cabeza, Analyst, 123(12) (1998) 2871. [19] R.Herraez Hernandez, P.Campins Falcó, A.Sevillano Cabeza, Anal.Chem., 68(5) (1996) 734. [20] D.Shangguan, Y.Zhao, H.Han, R.Zhao, G.Liu, Anal. Chem., 73 (2001) 2054. [21] S. Meseguer-Lloret, C. Molins-Legua, P. Campins-Falcó, J. of Chromatogr. A, 978 (2002) 59. [22] C.Molins Legua, P.Campíns Falcó, S. Meseguer Lloret, Chromatographia, 58 (2003) 15. [23] C.Dodeigne, L.Thunus, R.Lejeune, Talanta, 51 (2000) 415. [24] T.Kawasaki, M.Maeda, A.Tsuji, J.Chromatogr., 328 (1985) 121. [25] K.Nakashima, M.Maeda, A.Tsuji, J.Chromatogr., 530 (1990) 154. [26] S.R.Spurlin, M.M.Cooper, Anal. Lett., 19 (1986) 2277. [27] J.Ishida, N.Horike, M. Yamaguchi, Anal.Chim.Acta, 302 (1995) 61. [28] H.Yoshida, R.Nakao, T.Matsuo, H.Nohta, M.Yamaguchi, J. Chromatogr. A. 907 (2001) 39. [29] J.B.Noffsinger, N.D.Danielson, Anal. Chem., 59 (1987) 865. [30] X.Wang, D.R. Bobbitt, Talanta, 53 (2000) 337. [31] Andrew W. Knight, Trends in Anal.Chem., 18 (1999) 47. [32] M.Cobo, M.Silva, J.Chromatogr. A, 848 (1999) 105. [33] C.Molins Legua, P.Campins Falcó, A.Sevillano Cabeza, Anal.Chim.Acta, 378 (1999) 83. [34] P.J.M.Kwakman, H. Koelewijn, I.Kool, U.A.T. Brinkman, G.J.De Jong, J.Chromatogr,. 511 (1990) 155.
- 186. REFERENCIAS BIBLIOGRÁFICAS 162 [35] M.Tod, J.Y.Legendre, J.Chalom, H.Kouwtli, M-Poulou, R.Farinotti, G.Mahuzier, J.Chromatogr., 594 (1992) 386. [36] S. Meseguer-Lloret, C.Molins-Legua, J.Verdú-Andrés, P.Campíns-Falcó, J. of Chromatogr. A, in press. [37] Y. Moliner-Martínez, C. Molins-Legua, P. Campíns-Falcó, Talanta, 62 (2004) 373. [38] R. Weinberger, J. of Chromatogr., 314 (1984) 155. [39] N. Hanoaka, J. of Chromatogr., 503 (1990) 155. [40] Directiva Europea 98/83/CEE. [41] Díaz-Santos S.A. Métodos Normalizados para el análisis de agues potables y residuales. 17 Ed.1992. Apha-Awwe-WPCF [42] H.Hara, A.Motoike, S.Okazaki, Anal.Chem., 59(15) (1987) 1995. [43] H.Hara, A.Motoike, S.Okazaki, Analyst, 113(1) (1988) 113. [44] F.Cañete, A.Ríos, M.D.Luque de Castro, M.Valcárcel, Analyst, 113 (1988) 739. [45] M.Menezes-Santos, B.Freire-dos-Reis, H.Bergamin, N.Baccan, Anal.Chim.Acta., 261(1-2) (1992) 339. [46] H.Shen, T.J.Cardwell, R.W.Cattrall, Analyst, 123 (1998) 2181. [47] Z.Genfa, T.Uehara, P.K.Dasgupta, A.D.Clarke, W.Winiwarter, Anal. Chem., 70 (1998) 3656. [48] Y.Huang, Shi-fen Mou, J.M.Riviello, J. of Chromatogr. A., 868(2) (1999) 209. [49] J.Li, P.K.Dasgupta, Z.Genfa, Talanta, 50 (1999) 617. [50] W.Qin, Z.Zhang, B.Li, Y.Peng, Talanta, 48 (1999) 225. [51] J.P.Hart, A.K.Abass, D.C.Cowell, A.Chappel, Electroanalysis, 11(6) (1999) 406. [52] A. Walcarius, V.Vromman, J.Bessiere, Sens-Actuators-B, B56(1-2) (1999) 136. [53] H.Mana, V.Spohn, Fres. J. Anal. Chem., 366 (2000) 825. [54] Schwarz, H. Kaden, G.Pausch, Fres. J. Anal. Chem., 367 (2000) 396. [55] S. Meseguer Lloret, C. Molins Legua, P. Campins Falcó, Intern. J. of Environ. Anal. Chem., 82 (2002) 475. [56] W.J. Youden, Anal. Chem.,19 (1947) 946. [57] P.Campins-Falco, J. Verdú-Andrés, F. Bosch-Reig, C.Molins-Legua, Anal. Chim. Acta, 302 (1995) 323. [58] Instruction manual for ammonia electrode (NH3) (Mettler Toledo GmbH, Process, Urdorf). [59] A. Aminot, R.Kerouel, D.Birot, Wat.Res. 35-7 (2001) 1777. [60] J.P. Hart, A.K. Abass, D.C. Cowell, A. Chappel, Electroanalysis, 11(1999) 406. [61] J. Schwarz, H. Kaden, G. Pausch, Fres. J. Anal. Chem., 367 (2000) 396. [62] M-Mori, K.Tanaka, M.I.H.Helaleh, Q.Xu, M.Ikedo, Y.Ogura, S.Sato, W.Hu, K.Hasebe, J.Chromatogr. A, 997 (2003) 191. [63] D.H.Thomas, M.Rey, P.E.Jackson, J.Chromatogr. A, 956 (2003) 181. [64] H.Mana, V.Spohn, Fres. J. Anal. Chem., 366 (2000) 825. [65] J.Li, P.K.Dasgupta, Anal.Chim.Acta, 398 (1) (1999) 33. [66] W.Qin, Z.J.Zhang, B.X.Li, Y.Y.Peng, Talanta, 48 (1999) 225. [67] M.Marcé, D.S.Brown, T.Capell, X.Figueras, A.F.Tiburcio, J. of Chromatogr. B: Biomed. Sci. and Appl., 666 (2) (1995) 329. [68] M.Cobo, M.Silva, J. Chromatogr. A, 848 (1999) 105. [69] P.J.M.Kwakman, D.A.Kamminga, U.A.Th.Brinkman, J. Chromatogr. 553 (1991) 345.
- 187. Capítulo 2. RESULTADOS OBTENIDOS Y DISCUSIÓN 163 [70] E.Orejuela, M.Silva, J. Chromatogr. A, 1007 (2003) 197. [71] C.Molins-Legua, P.Campins-Falco, A.Sevillano-Cabeza. Anal. Chim. Acta, 378 (1999) 83. [72] D.L. Massart, B.G.M.Vandeginste, L.M.C.Buydens, S.De Jong, P.J.Lewi, J.Smeyers-Verbeke, Handbook of Chemometrics and Qualimetrics: Part A, Elsevier, Amsterdam 1997. [73] M. Valcárcel, S.Cárdenas, M.Gallego. Trends Anal. Chem., 18 (1999), 685. [74] M. Valcárcel, S.Cárdenas, M.Gallego, Trends Anal. Chem., 21 (2002), 251. [75] S.Igarashi, T.Nagoshi, T.Kotake, Anal. Letters, 33 (2000), 3271. [76] L.A. Tortajada-Genaro, P.Campíns-Falcó, J.Verdú-Andrés, F.Bosch-Reig, Anal. Chim. Acta, 450 (2001) 155. [77] S.Meseguer-Lloret, P.Campíns-Falcó, L.A.Tortajada-Genaro, F.Blasco-Gómez, Intern. J. of Environ. Anal. Chem., 83 (2002) 405. [78] Y. Moliner-Martínez, S.Meseguer-Lloret, L.A.Tortajada-Genaro, P.Campíns-Falcó, Talanta, 60 (2002) 257. [79] N.W. Barnett, R. Bos, R.N. Evams, R.A. Russell. Anal. Chim. Acta, 403 (2000) 145. [80] S. Igarashi, T. Nagoshi, T. Kotake, Anal. Letters, 33 (2000) 3271. [81] M.A. Marina-Sánchez, M.E. Díaz-García, A. Sanz-Mendel. Mikrochim-Acta, 106 (1992) 227. [82] R. Escobar, Q.X. Lin, A. Guiraum, F.F. De la Rosa. Analyst, 118 (1993) 643. [83] HG. Beere, P. Jones, Anal. Chim. Acta, 293 (1994) 237. [84] R. Escobar, Q. Lin, A. Guiraum, F.F. De la Rosa. Inter. J. Environ. Anal. Chem., 61 (1994) 169. [85] B.Gammelgaard, Y.P. Liao, O. Jons, Anal.Chim.Acta, 354 (1997) 107. [86] A. Economou, A.K. Clark, P.R.Fielden, Anal-Commun., 35 (1998) 389. [87] M. Derbyshire, A. Lamberty, P.Gardiner, Anal. Chem., 71 (1999) 4203. [88] Y. Xu, F.G. Bessoth, JCT Eijkel and A. Manz, Analyst, 125 (2000) 677. [89] A.E. Greenberg, L.C. Clesceri, A.D. Eaton, Standard Methods for Examination of Water and Wastewaters, American Public Health Association, Washington D.C., (1992). [90] M. Stoeppler. Sampling and Sample preparation, Elsevier, Berlin, 1997. [91] E. Nakayama, K. Isshiki, Y. Sohrin and H. Karatami, Anal. Chem., 61 (1989) 1392. [92] A.A.Alwarthan, K.A.J. Habib, A. Townshend, Fres. J. Anal. Chem., 337 (1990) 848. [93] B. Yan, P.J. Worsfold, Anal. Chim. Acta, 236 (1990) 287. [94] V.A. Elron, K.S. Johnson, K.H. Coale, Anal. Chem., 63 (1991) 893. [95] H. Obata, H. Karatani, E. Nakayama, Anal. Chem., 65 (1993) 1524. [96] Z. Zhang, W. Qin, S. Liu, Anal. Chim. Acta, 318 (1995) 71. [97] S. Hirata, Y. Hashimoto, M. Aihara, G. Vitharana-Mallika, Fres. J. Anal. Chem., 355 (1996) 676. [98] K. Okamura, T. Gamo, H. Obata, E. Nakayama, Y. Nozaki, Anal. Chim. Acta, 377 (1998) 125. [99] W. Qin, Z.J. Zhang, F.C. Wang, Fres. J. Anal. Chem., 360 (1998) 130. [100] A.R. Bowie, E.P. Achterberg, R.F.C. Mantoura, P.J. Worsfold, Anal. Chim. Acta, 361 (1998) 189. [101] S. Hirata, H. Yoshihara, M. Aihara, Talanta, 49 (1999) 1059. [102] V. Cannizzaro, A.R. Bowie, A. Sax, E.P. Achterberg, P.J. Worsfold, Analyst, 125 (2000) 51.
- 188. REFERENCIAS BIBLIOGRÁFICAS 164 [103] C.A. Chang, H.H. Patterson, Anal. Chem., 52 (1980) 653. [104] Q.X. Lin, A. Guirarum, R. Escobar, F.F. de la Rosa, Anal. Chim. Acta., 283 (1993) 379. [105] A.R. Bowie, P.R. Fielden, R.D. Lowe, R.D. Snook, Analyst, 120 (1995) 2119. [106] Y. Zhou, G. Zhu, Talanta, 44 (1997) 2041. [107] H. Zamrow, K.H. Coale, K.S. Johnson, C.M. Sakamoto, Anal. Chim. Acta, 377 (1998) 133. [108] P. Campíns-Falcó, L.A. Tortajada-Genaro, F. Bosch-Reig, Talanta, 55 (2001) 403. [109] F.W.Fifield, P.J.Haines, Environ. Anal. Chem., Blackie, London, 1995. [110] N. Shakulashvili, T. Faller and H. Engelhardt, J. Chromatogr. A, 895 (2000) 205. [111] W. Zmijewska, H. Polowska-Motrenko, H. Stokowska, J. Radioanal. Nucl. Chem., 116 (1987) 243. [112] N.S. Chong, M.L. Norton, J.L. Anderson, Anal. Chem., 62 (1990) 1043. [113] M.A. Barreiros, M.L. Carvalho, M.M. Costa, M.I. Marques, M.T. Ramos, X Ray Spectrom., 26 (1997) 165. [114] R. Saran, T.S. Basu-Baul, P. Srinivas, D.T. Khalthing, Anal. Lett., 25 (1992) 1545. [115] T. Balaji, P. Chiranjeevi, G.R.K. Naidu, Anal. Lett., 31 (1998) 1081. [116] B. Budic, Fres. J. Anal. Chem., 368 (2000) 371. [117] K.H. Lee, M. Oshima, T. Takayanagi, S. Motomizu, Anal. Sci., 16 (2000) 731 [118] J.R. Gord, G. Gordon, G.E. Pacey, Anal. Chem., 60 (1988) 2. [119] D.F. Marino, J.D. Ingle, Anal. Chem., 53 (1981) 292. [120] H. Sakai, T. Fujiwara, M. Yamamoto and T. Kumamaru, Anal. Chim. Acta, 221 (1989) 249. [121] R. Ferrer, J.L. Beltrán, J. Guiteras, Talanta, 45 (1998) 1073. [122] J. Amador-Hernández, A. Cladera, J.M. Estela, P.L. López-Alba, V. Cerda, Analyst, 123 (1998) 2235. [123] F. Blasco-Gómez, F. Bosch-Reig, P. Campíns-Falcó, Talanta, 49 (1999) 155. [124] M. Azubel, F.M. Fernández, M.B. Tudino, O.E. Troccoli, Anal. Chim. Acta, 398 (1999) 93. [125] P. Campins-Falcó, L.A. Tortajada-Genaro, S. Meseguer-Lloret, F. Bosch-Reig, Anal. Bioanal. Chem., 374 (2002) 1223. [126] D. González-Robledo, M.Silva, D.Pérez-Bendito, Anal. Chim. Acta, 217 (1989) 239. [127] P. Campins-Falcó, L.A.Tortajada-Genaro, F.Bosch-Reig, Talanta, 55 (2001) 403. [128] R.D. Jalkian, M.B. Denton, Appl. Spectrosc., 42 (1998) 1194. [129] A. Segura-Carretero, J. Rodríguez-Fernández, A.R. Bowie, P.J. Worsfold, Analyst, 125 (2000) 387. [130] T.P. Burt, A.L. Heathwaite, S.T. Trudgill. Nitrate: Processes, Patterns and Management, John Wiley & Sons, New York, 1993. [131] E.-S. A. Badr, E.P. Achterberg, A.L. Tappin, S.J. Hill, C.B. Braungardt, Trends Anal. Chem., 22 (2003) 819. [132] K.Cameron, C.Madramootoo, A.Crolla, C.Kinsley, Wat. Res., 37 (2003) 2803. V. P. Aneja, B. Bunton, J. T. Walker, B. P Malik, Atm. Environ., 35 (2001) 1949. [133] S.E.Tsiouris, A.P.Mamolos, K.L.Kalburtji, D.Alifrangis, Agric. Ecosyst. & Envir., 89 (2002) 117. [134] S.H. Ensign, M.A. Mallin, Wat. Res., 35 (2001) 3381.
- 189. Capítulo 2. RESULTADOS OBTENIDOS Y DISCUSIÓN 165 [135] V. P. Aneja, B. Bunton, J. T. Walker, B. P Malik, Atm. Environ., 35 (2001) 1949. [136] B.M. Jones, C.G. Daughton, Anal. Chem., 57 (1985) 2320. [137] EPA-821-R-01-004. Method, 1687. 2001. [138] G. Alavoine, B. Nicolardot, Analusis, 28 (2000) 88. [139] J.H. Sharp, K.R. Rinker, K.B. Savidge, J.Abell, J.Y. Benaim, D. Bronk, D.J. Burdige, G. Cauwet, W. Chen, M.D. Doval et al., Mar. Chem., 78 (2002) 171. [140] A. Aminot, D.S. Kirkwood and R. Kérouel, Mar.Chem., 56 (1997) 59. [141] M. Kaminski, D.Jastrzebski, A.Przyjazny, R.Kartanowicz, J.Chromatogr. A, 947 (2002) 217. [142] F. Dai, V.P. Burkert, H. N. Singh, W.L. Hinze, Microchemical Journal, 57 (1997) 166. [143] P. Campíns-Falcó, C. Molins-Legua, A. Sevillano-Cabeza and L. A. Tortajada Genaro, J. Chromatogr. B, 759 (2001) 285. [144] H.Mana, U.Spohn, Fres. J. Anal. Chem., 366 (2000) 825. [145] A.Sevillano Cabeza, Y.Moliner Martinez, C.Molins Legua, P.Campíns Falcó, Anal.Bioanal.Chem., 376 (2003) 918. [146] J. Verdú, D.L. Massart, Applic. Spectrosc., 52 (1998) 1425. [147] V.Centner, D.L.Massart, O.E. de Noord, S.de Jong, B.M.Vandeginste and C.Stena. Anal.Chem., 68 (1996) 3851. [148] J. Li, P. Purnendu, K. Dasgupta, Anal. Chim. Acta, 398 (1999) 33. [149] D. Tang, L.Wen, P.H. Santschi, Anal. Chim. Acta, 408 (1-2) (2000) 299.
- 192. 168
- 193. Capítulo 3. CONCLUSIONES FINALES 169 3. CONCLUSIONES FINALES Los trabajos expuestos pretenden aportar soluciones a algunos problemas planteados en el análisis de aguas. Se proponen técnicas con diferentes grados de automatización, utilizando diferentes fuentes de señal y aplicadas a diferentes analitos y a una amplia variedad de muestras de agua. Estas características, así como los métodos de validación empleados, y los pasos analíticos cuyo estudio se ha resaltado en mayor medida, se reflejan en la Tabla 77. Tabla 77. Principales características de los métodos propuestos en esta Tesis. Aminas–DnsCl-off Aminas–DnsCl-on Aminas–DnsCl-CL NH4 + -OPA/NAC NH4 + -DnsCl-CL Cu(II)-CL Cr(III)/Cr(VI) Cr(III)/Co(II)/Cu(II) Acondicionamiento Cr(III)/Co(II)-PLS NH4 + -aminas/ OPA-NAC Norgánicoamino+ NH4 + -Luminol(CL) Estático X X X X Flujo X X X X HPLC-on X Grado de automatización HPLC-off X X X Derivatización X X X X X X X Efecto catalítico X X XFuente de señal Quenching X Cinética X X X XSeñal frente al tiempo Flujo X X X X Orgánico X X X X X Analito Inorgánico X X X X X X X X Agua de Riego 1 X X X X X Agua de Fuente X X X X X Agua Residual X X X X X X X Agua de Grifo X X X X X Agua de Riego 2 X X X Agua de Lago 1 X Agua de Lago 2 X X X Agua Natural 1 X X X Agua Natural 2 X X X Agua Natural 3 X Agua Marina 1 X Agua Marina 2 X Muestras de agua Agua Marina 3 X X X Preconcentración X X X Selección Modelo Calibración X X X Interferencias X X X X X X X Derivatización X X X X X X Proced. Kjeldahl X X X Separación HPLC X X X X R. post-columna X X Estudio específico de un paso analítico Posible Screening X X X X X
- 194. Capítulo 3. CONCLUSIONES FINALES 170 %Recuperación X X X X X X X Estudio de fuentes de error X X Mét. Referencia X X X MRC X X X X Validación del método Muestra sintética X En cuanto al grado de automatización, se han desarrollado métodos en estático con detección QL para la determinación de iones metálicos, o fluorescentes para la determinación simultánea de amonio y grupos amino primario; métodos en flujo con detección QL para la detección de iones metálicos o del Nitrógeno orgánico amino + amoniacal; y métodos cromatográficos fuera de línea (off-line) con detección fluorescente, UV o QL para la detección de aminas o amonio, y en línea (on-line) con detección fluorescente para la detección de aminas. La fuente de señal normalmente la proporcionó un derivado del analito, como el derivado dansilado o el isoindol derivado de OPA/NAC. En la reacción la determinación del Nitrógeno orgánico amino más amoniacal, el derivado formado es la cloramina, y el efecto en la señal es la disminución por eliminación del hipoclorito. En otros casos, la señal la proporcionó el intermedio de reacción (3-aminoftalato en estado excitado) formado gracias a la presencia del analito que actuaba como catalizador. Este es el caso de los iones metálicos Cr(III), Co(II) y Cu(II) en la reacción del luminol. Otro tipo de señal medida ha sido el efecto quenching causado por el analito sobre la reacción quimioluminiscente (el analito absorbe la luz emitida), como en el capítulo de la determinación QL del Cu(II). Ha habido dos tipos de variación de señal frente al tiempo: al medir la cinética de la reacción, o al hacer las medidas de inyección en flujo. Por otra parte, las medidas cromatográficas también se registraron en función del tiempo de retención. Se han analizado varios tipos de analitos: orgánicos (aminas alifáticas y poliaminas, Nitrógeno Orgánico amino) e inorgánicos (Cr(III), Co(II), Cu(II), NH4 + ). Además se han analizado un amplio rango de muestras de agua, abarcando una pequeña parte de la gran variedad que podemos encontrar: Agua de riego (1 y 2 corresponden a dos tomas de muestra en dos puntos de muestreo y dos temporadas diferentes), agua de fuente, agua residual (procedente de una industria de pesticidas), agua de grifo, agua de lago (procedente del Lago de La Albufera de Valencia, 1 y 2 corresponden a dos tomas de muestra en dos puntos de muestreo y dos estaciones diferentes), agua natural (son aguas de consumo humano: 1, 2 y 3 corresponden a diferentes marcas comerciales) y agua marina (procedente de la costa de Valencia: 1, 2 y 3 corresponden a tres tomas de muestra en tres puntos de muestreo y dos estaciones diferentes). Los principales pasos analíticos cuyo estudio se ha resaltado específicamente han sido: la preconcentración de los analitos en el caso de la determinación de aminas alifáticas; la selección del modelo de calibración en el estudio de la determinación quimioluminiscente de Cromo o en el estudio de la determinación simultánea de amonio y grupos amino primario utilizando el método de OPA/NAC; el estudio de los posibles
- 195. Capítulo 3. CONCLUSIONES FINALES 171 interferentes en la mayoría de los métodos propuestos; el estudio de las reacciones de derivatización con Cloruro de dansilo o con OPA/NAC según el caso; el estudio de la reacción QL post-columna en los capítulos de separación cromatográfica de los derivados dansilados de aminas y de amonio; el tratamiento Kjeldahl de digestión de las muestras en el caso de la medida de concentraciones totales de Nitrógeno orgánico amino y amonio; y la posibilidad de adaptar el método para el screening de muestras como en el capítulo de la determinación QL de Cu(II). Por último, los métodos propuestos se han validado utilizando diferentes procedimientos: mediante estudios de recuperación sobre muestras reales, utilizando métodos de referencia, materiales de referencia certificados, muestras sintéticas o mediante el estudio de las fuentes de error, obteniéndose resultados exactos. Las principales ventajas analíticas que presentan los métodos propuestos se reflejan en la Tabla 78. Tabla 78. Ventajas de los métodos propuestos (a mayor número de asteriscos *, mejor condición). Aminas - DnsCl - off Aminas - DnsCl - on Aminas - DnsCl - CL NH4 + - OPA/NAC NH4 + - DnsCl - CL Cu (II) - CL Tratamiento de la muestra Preconc en C18 Preconc en C18 Preconc en C18 Ajustar pH Ajustar pH Ajustar pH Bajo consumo de reactivos ** ** ** *** ** ** Instrumentac. simple ** ** ** *** ** ** Selectividad Si Si Si Si, a λexc = 415 nm Si Si Precisión DER < 4 % (EA) DER < 11.7% (EA) DER < 10% (EA) DER < 12.5% DER < 8.6% DER < 16% Exactitud Er = 7 % Er = 11% Er = 10.7% Er < 11% Er < 9.8% %Rec = 109% Límite de detección 2 µg/L (EA) 15 µg/L (EA) 0.2 µg/L (EA) 70 µg/L NH4 + 8 µg/L NH4 + 9 µg/L Intervalo Lineal 0.01-0.5 mg/L (EA) 0.5-2.25 mg/L (EA) 0.7-64 µg/L (EA) 0.25 – 1.4 mg/L NH4 + 0.027 – 0.645 mg/L NH4 + 9-100 µg/L Cu Velocidad de muestreo * * * ** * **
- 196. Capítulo 3. CONCLUSIONES FINALES 172 Tabla 78 (continuación). Ventajas de los métodos propuestos (a mayor número de asteriscos *, mejor condición). Cr(III)/ Cr(VI) Cr(III)- Co(II)-Cu(II) Acondicionam. Cr(III)/Co(II) - PLS NH4 + -aminas/ OPA-NAC N amino + NH4 + / Luminol Tratamiento de la muestra Reducción Acondic. ácidos No Ajustar pH Ajustar pH Bajo consumo de reactivos ** ** *** *** ** Instrumentac. simple ** ** *** *** ** Selectividad Si, después reducción Si, acondic. Si Si Si Precisión DER < 12% - - - DER < 3.7 % Exactitud Er < 7% - - Er NH4+ < 24% Er MA < 8% %Rec ≈100% Límite de detección 2 µg/L 2.2 µg/L Cr 0.5 µg/L Co 2.1 µg/L Cu - 70 µg/L NH4 + 38 µg/L MA 70 µg/L N Intervalo Lineal 3-10 µg/L 3-15 µg/L Cr 2-8 µg/L Co 10-50 µg/L Cu 0-80 µg/L Cr 0-80 µg/L Co 0.25-1.75 mg/L NH4 + ; 0.25-0.625 mg/L MA 0.25-4 mg/L N Velocidad de muestreo *** *** *** ** *** En la esta tabla, se observa que el tratamiento realizado sobre las muestras para la aplicación de los métodos propuestos ha sido mínimo, tanto en métodos quimioluminiscentes como en métodos fluorescentes o UV. El tratamiento realizado ha incluido etapas sencillas como la reducción de Cr(VI) a Cr(III) en el punto 2.2.1.1 o la preconcentración en cartuchos de extracción en fase sólida en los puntos 2.1.1.1 y 2.1.1.3 de derivatización de aminas, requiriendo los demás métodos únicamente el ajuste del pH de las muestras. El consumo de reactivos fue menor y la instrumentación más sencilla en los métodos estáticos respecto a los métodos en flujo o cromatográficos, pero a cambio en los métodos estáticos se pierde el grado de automatización. Por su sencillez, hemos de destacar el detector quimioluminiscente utilizado en el capítulo de determinación y screening de muestras para Cu(II). Además cabe resaltar la ventaja en la sencillez de la instrumentación en los métodos quimioluminiscentes, puesto que los sistemas de detección utilizados no requirieron fuente de excitación. En cuanto a la selectividad, los métodos propuestos resultaron selectivos para la medida de los analitos en estudio, tanto las técnicas fluorescentes y UV, como las quimioluminiscentes. En cuanto a las medidas de exactitud y precisión, la desviación estándar relativa así como los errores relativos fueron adecuados, y similares en todas las técnicas estudiadas.
- 197. Capítulo 3. CONCLUSIONES FINALES 173 Los métodos quimioluminiscentes utilizados destacan sobre los fluorescentes o UV, en los límites de detección e intervalos lineales alcanzados. En la determinación del N Total, los límites de detección obtenidos fueron similares utilizando la reacción fluorescente del OPA/NAC o la reacción QL del Luminol. Sin embargo, si se comparan los límites de detección para los estudios de aminas alifáticas, se observa que la detección quimioluminiscente proporcionó límites de detección del orden de 10 veces inferior. Lo mismo ocurre al comparar los límites de detección obtenidos utilizando el método fluorescente OPA/NAC o del método QL de los derivados dansilados-TCPO-H2O2 para la determinación de amonio. El límite de detección alcanzado mediante la detección QL es del orden de 10 veces inferior respecto a la detección fluorescente. Esta sería pues una de las principales ventajas de la quimioluminiscencia. La velocidad de muestreo fue mayor al utilizar métodos en flujo que no requerían tiempo de reacción, seguida de los métodos estáticos de tiempo de reacción corto, y por último los métodos cromatográficos que incluían una etapa de derivatización además de la separación cromatográfica. Los métodos propuestos para la determinación de aminas alifáticas presentan ventajas respecto a los existentes en la bibliografía. En primer lugar, el uso de cartuchos C18 de extracción en fase sólida para la preconcentración y derivatización de los analitos, proporcionó buena sensibilidad en unas condiciones de reacción sencillas y rápidas. Al automatizar el proceso, solamente para algunas de las aminas en estudio, se mejoró la sensibilidad, disminuyendo para otras, pero se ganó en grado de automatización de la técnica. Por último, al acoplar el módulo de reacción post-columna (sistema off-line, detección quimioluminiscente), se mejoró la sensibilidad en todos los casos, y si se comparan los límites de detección alcanzados mediante esta técnica, con los publicados en la bibliografía reciente para otros métodos fluorimétricos o UV (Tabla 5, Apéndice 3), se observa que los límites de detección alcanzados son mejores. El método fluorimétrico de OPA/NAC propuesto para la determinación de amonio presentó ventajas frente al método de referencia de Nessler y el método del electrodo selectivo (Tabla 8, Apéndice 4). El intervalo dinámico de concentraciones fue más corto, pero el límite de detección alcanzado fue menor, del orden de 0.07 mg/L. La reproducibilidad fue similar para los tres métodos, el método del electrodo selectivo presentó interferencia de aminas con señal no aditiva, mientras que el método de Nessler y el de OPA/NAC a λexc = 415 nm, fueron selectivos para amonio. En cuanto a los costes, el método del electrodo selectivo era el más barato, y en cuanto a la rapidez, la velocidad de muestreo fue mayor para los métodos del electrodo selectivo y de OPA/NAC. El método quimioluminiscente propuesto posteriormente, además de mejorar la sensibilidad y selectividad en la determinación de amonio, es novedoso, pues aunque la reacción entre los compuestos de nitrógeno y el cloruro de dansilo es bien conocida, esta reacción no se había aplicado todavía a la determinación de amonio. La marcada tendencia de la Química Analítica hacia el desarrollo de sistemas de screening, cuyas características de sencillez en la instrumentación, y rapidez en la respuesta los hacen óptimos para seleccionar entre un conjunto de muestras, aquellas
- 198. Capítulo 3. CONCLUSIONES FINALES 174 cuyo contenido en un determinado analito sea superior a un nivel determinado (el marcado por la legislación, el cliente, etc.), se refleja en el trabajo desarrollado en el Apéndice 6, en el que se desarrolló un sistema de screening de muestras para Cu(II). En este trabajo, no se pretende alcanzar la máxima sensibilidad posible mejorando los trabajos presentados en la bibliografía, sino presentar un sistema de screening con las características adecuadas para discriminar entre un conjunto de muestras cuyo contenido de Cu(II) esté por encima o por debajo del límite legislado (50 µg/L) en buenas condiciones de reproducibilidad y fiabilidad. Al aplicar la reacción quimioluminiscente del luminol en la especiación de Cromo, se alcanzó la misma sensibilidad que con otros métodos (espectrofotometría, fluorimetría, fluorescencia de rayos X, espectrometría de absorción atómica, espectrometría de emisión atómica, cromatografía y métodos electroquímicos entre otros) utilizando esta técnica de menor coste. Se puede utilizar el valor de la pendiente del MOSA para garantizar la ausencia de errores del método dada su robustez. Por otra parte, en la bibliografía no existen referencias que estudien la influencia de las condiciones de acondicionamiento de las muestras en la señal quimioluminiscente. El estudio de la señal quimioluminiscente directa producida por la reacción del luminol-peróxido de hidrógeno catalizada por iones metálicos como Cr(III), Co(II), Fe(II), Fe(III), Ni(II) y Mn(II) en función del protocolo de acondicionamiento de la muestra (sin acidificar o acidificadas con HCl o HNO3, en presencia y ausencia de EDTA, en estático y en flujo), permitió seleccionar las condiciones para la determinación selectiva de Cr(III) o para la determinación total de Cr(III), Co(II) y Cu(II). Además por primera vez se han aplicado los perfiles intensidad-tiempo descritos por la reacción del luminol/H2O2 catalizada por Cromo o Cobalto en estático, en calibración multivariada para el análisis quimioluminiscente simultáneo de dos especies. El método OPA/NAC permitió la determinación simultánea de amonio y grupos amino primario por calibración bivariada o multivariada libre de errores sistemáticos con resultados similares, siendo un buen indicador de cuándo el tratamiento Kjeldahl falla. El método quimioluminiscente para la determinación del nitrógeno orgánico amino y amoniacal, permitió utilizar una misma curva de calibrado para un gran número de compuestos de nitrógeno. El Nitrógeno orgánico disuelto y el amonio pueden deducirse de forma muy rápida, y para algunos tipos de muestras, el tratamiento Kjeldahl se puede sustituir por el sistema de inyección en flujo presentado. Esta opción es muy ventajosa, puesto que la toxicidad de los reactivos es baja y el método se puede utilizar para monitorización in situ. La metodología propuesta también puede utilizarse como alternativa para la detección final del amonio después del tratamiento de digestión Kjeldahl. Resumiendo, y de acuerdo con los objetivos específicos marcados en la Pág. 4, esta Tesis contribuye a: 1. Mejorar el análisis de screening de aminas alifáticas en muestras de agua de diferente naturaleza y disminuir sus límites de detección.
- 199. Capítulo 3. CONCLUSIONES FINALES 175 2. Disminuir los límites de detección de amonio en aguas e incrementar la exactitud de su determinación. 3. Contribuir al conocimiento de los métodos de screening de muestras. 4. Mejorar la exactitud de la determinación de algunos iones metálicos controlando los errores sistemáticos y la especiación de Cr(VI) y Cr(III). 5. Contribuir al estudio del Nitrógeno Orgánico disuelto. Y todo ello teniendo como marco el profundizar en el desarrollo de los métodos quimioluminiscentes.
- 200. Capítulo 3. CONCLUSIONES FINALES 176 SUGERENCIAS PARA TRABAJOS FUTUROS Sin duda la continuidad del trabajo realizado en esta Tesis, seguiría las tendencias actuales de la Química Analítica. En el caso de la derivatización de aminas o de amonio con separación cromatográfica y reacción quimioluminiscente postcolumna, la siguiente fase sería la automatización completa del sistema, minimizando así los errores experimentales debidos al operador. También existe una marcada tendencia en Química Analítica hacia la miniaturización de sistemas, que aportaría las ventajas del mínimo consumo tanto de reactivos como de muestra. Esta sería otra de las propuestas tanto para la detección de Cr/Co utilizando las reacción del Luminol/H2O2, como para la detección del Cu(II) utilizando la reacción del TCPO/H2O2/Coproporfirina I. También cabría la posibilidad de la miniaturización de los sistemas cromatográficos para la detección de aminas alifáticas o amonio, tema prioritario en el grupo para el que se ha conseguido financiación (BQU2003-6138). Por último, el diseño de sensores es otro campo de interés en crecimiento. Existe una ausencia de sensores adecuados para la monitorización en tiempo real y en línea. En este sentido el desarrollo de sensores quimioluminiscentes sería otra alternativa para trabajos futuros, tendiendo a una mejora en cuanto a las propiedades de emplazamiento, principio de muestreo, tratamiento de la muestra, número de medidas, necesidad de suministros o intervalo de servicio.
- 201. 177 4. APÉNDICES
- 202. 178
- 203. Capítulo4. APÉNDICES 179 APÉNDICES Apéndice 1. S. Meseguer-Lloret, C.Molins-Legua, P. Campíns-Falcó. Departament de Química Analítica, Facultat de Química, Universitat de Valencia. Preconcentration and dansylation of aliphatic amines using C18 solid-packings. Application to the screening analysis in environmental water samples. J. of Chromatography A, 978 (2002) 59-69. Apéndice 2. C.Molins-Legua, P. Campíns-Falcó, S. Meseguer-Lloret. Departament de Química Analítica, Facultat de Química, Universitat de Valencia. Derivatization on solid supports: An alternative method for solution derivatization of amines in several matrices. Chromatographia, 58 (2003) 15-27. Apéndice 3. S. Meseguer-Lloret, C.Molins-Legua, J.Verdú-Andrés, P. Campíns-Falcó. Departament de Química Analítica, Facultat de Química, Universitat de Valencia. Sensitive determination of aliphatic amines in water by High-Performance Liquid Chromatography with chemiluminescence detection. J. of Chromatography A, en prensa. Available on line at www.sciencedirect.com. Apéndice 4. S. Meseguer-Lloret, C.Molins-Legua, P. Campíns-Falcó. Departament de Química Analítica, Facultat de Química, Universitat de Valencia. Ammonium determination in water samples by using OPA-NAC reagent: A comparative study with Nessler and Ammonium Selective Electrode methods. Intern. J. of Environ. Anal. Chem. 82 (7) (2002) 475-489. Apéndice 5. S. Meseguer-Lloret, C.Molins-Legua, P. Campíns-Falcó. Departament de Química Analítica, Facultat de Química, Universitat de Valencia. Selective chemiluminescence determination of ammonium in water by HPLC. Enviado a J. of Chromatography A. Apéndice 6. S. Meseguer-Lloreta , P. Campíns-Falcóa , S. Cárdenasb , M. Gallegob , M. Valcárcelb . a Departament de Química Analítica, Facultat de Química, Universitat de Valencia. b Departamento de Química Analítica, Campus de Rabanales, Universidad de Córdoba. FI automatic method for the determination of copper(II) based on coproporphyrin I - Cu(II) / TCPO / H2O2 chemiluminescence reaction for the screening of waters. Talanta, revised paper (minor revision).
- 204. Capítulo4. APÉNDICES 180 Apéndice 7. S. Meseguer-Lloret, P. Campíns-Falcó, L. A. Tortajada-Genaro, F. Blasco Gómez. Departament de Química Analítica, Facultat de Química, Universitat de Valencia A guide to avoid method bias of Chromium (III, VI) chemiluminescence determination by luminol-hydrogen peroxide reaction. Application to water samples. Intern. J. Environ. Anal. Chem. 83 (5) (2002) 405-416. Apéndice 8. Y. Moliner-Martínez, S. Meseguer-Lloret, L. A. Tortajada-Genaro, P. Campíns-Falcó. Departament de Química Analítica, Facultat de Química, Universitat de Valencia. Influence of water sample storage protocols in chemiluminescence detection of trace elements. Talanta 60 (2003) 257-258. Apéndice 9. P. Campíns-Falcó, L. A. Tortajada-Genaro, S. Meseguer-Lloret, F. Bosh-Reig. Departament de Química Analítica, Facultat de Química, Universitat de Valencia. Multivariate calibration applied to simultaneous determination of cobalt and chromium. Anal. Bioanal. Chem. 374 (2002) 1223-1229. Apéndice 10. S. Meseguer-Lloret, J.Verdú-Andrés, C.Molins-Legua, P. Campíns-Falcó. Departament de Química Analítica, Facultat de Química, Universitat de Valencia. Modified Roth’s fluorimetric method for water samples: ammonium and primary amine groups determination and utility in total nitrogen Kjeldahl. Enviado a Talanta. Apéndice 11. S. Meseguer-Lloret, C.Molins-Legua, J.Verdú-Andrés, P. Campíns-Falcó. Departament de Química Analítica, Facultat de Química, Universitat de Valencia. Chemiluminescence authomatic method for total estimation of organic nitrogen (-CH2- NH-) and ammonium. Enviado a Analytica Chimica Acta.
- 205. 181 Apéndice 1. S. Meseguer-Lloret, C.Molins-Legua, P. Campíns-Falcó. Departament de Química Analítica, Facultat de Química, Universitat de Valencia. Preconcentration and dansylation of aliphatic amines using C18 solid-packings. Application to the screening analysis in environmental water samples. J. of Chromatography A, 978 (2002) 59-69. Enlace URL: http://www.sciencedirect.com
- 206. 182
- 207. 183 Apéndice 2. C.Molins-Legua, P. Campíns-Falcó, S. Meseguer-Lloret. Departament de Química Analítica, Facultat de Química, Universitat de Valencia. Derivatization on solid supports: An alternative method for solution derivatization of amines in several matrices. Chromatographia, 58 (2003) 15-27. Enlace URL: http://www.springerlink.com
- 208. 184
- 209. 185 Apéndice 3. S. Meseguer-Lloret, C.Molins-Legua, J.Verdú-Andrés, P. Campíns-Falcó. Departament de Química Analítica, Facultat de Química, Universitat de Valencia. Sensitive determination of aliphatic amines in water by High-Performance Liquid Chromatography with chemiluminescence detection. J. of Chromatography A, en prensa. Enlace URL: http://www.sciencedirect.com
- 210. 186
- 211. 187 Apéndice 4. S. Meseguer-Lloret, C.Molins-Legua, P. Campíns-Falcó. Departament de Química Analítica, Facultat de Química, Universitat de Valencia. Ammonium determination in water samples by using OPA-NAC reagent: A comparative study with Nessler and Ammonium Selective Electrode methods. Intern. J. of Environ. Anal. Chem. 82 (7) (2002) 475-489.
- 212. 188
- 213. 189 Apéndice 5. UNPUBLISHED PAPER SELECTIVE CHEMILUMINESCENCE DETERMINATION OF AMMONIUM IN WATER BY HPLC S. Meseguer Lloret, C. Molins Legua and P.Campíns Falcó* Departament de Química Analítica, Facultad de Química, Universitat de Valencia C/ Dr. Moliner 50, E46100- Burjassot, Valencia. SPAIN ABSTRACT A selective and sensitive method has been developed for liquid chromatographic determination of ammonium in water samples. Analyte is solution pre-column derivatized with Dansyl Chloride. Optimal solution derivatization conditions have been established. The dansyl derivative is chromatographied and post column mixed with peroxyoxalate (TCPO) and H2O2 in order to perform chemiluminescence detection. Detection limit achieved is 8 µg/L and linear response from 0.027 to 0.750 mg/L of ammonium. Under optimum chromatographic conditions, ammonium ion was determined within 2.4 min. The method is fast, and near 10 derivatized samples can be measured per hour. No interference of amines has been obtained. Accuracy studies gave % relative error below 9.8%, and precision studies gave %RSD between 2-8%. The method has resulted reliable for amine and ammonium screening and quantification of ammonium in different kind of water samples: bottled, tap, lake, fountain, irrigation, residual and sea. Only matrix effect was found in sea water samples. The method was also applied to several samples after applying Kjeldahl treatment in order to obtain values of Total Kjeldahl Nitrogen. Keywords: ammonium, water samples, dansylation, post-column chemiluminescence, HPLC, Total Kjeldahl Nitrogen. * Correspondence author. E-mail address: pilar.campins@uv.es (P.Campins).
- 214. 190 INTRODUCTION Ammonium is one of the most widely used industrial chemicals. The principal uses of ammonia are direct-application fertiliser, conversion to other chemical fertilizers, fibres, explosives, plastics, and animal feeds. It is also used as food additive, in insecticides, in the manufacture of household detergents and cleaners, and as a major commercial refrigerant [1]. It is the most serious pollutant causing eutrophication of aqueous environments. Although ammonium is an important link in the nitrogen cycle of aquatic systems, its determination is still a delicate task. Its concentration in water systems is regulated by legislation to guarantee the quality of the water supplied for human consumption, being the limit (parametric value) established in 0.5 mg/L , and 2 mg/L when we are talking about Total Kjeldahl Nitrogen [2]. The performance characteristic of the method of analysis is also specified in the legislation, it has to be capable of measuring concentrations equal to the parametric value with trueness, precision and limit of detection of 10 % of the parametric value [2]. The reference methods for ammonium determination include wet technique such as trimetry, colorimetry (Nesslerization, Phenate or automated phenate) or ammonia- selective electrode [3]. These methods suffer from several chemical interferences, can require distillation step and often offer lack of sensitivity [4-7]. In order to improve selectivity some procedures have been described in the literature in which a ion chromatography was applied. In these papers a weakly basic anion-exchange resin column [8] or high capacity cation exchange column [9], have been used to separate ammonium to the other inorganic cations or/and aliphatic amines which can be present in water samples. A number of methods exist for measuring ammonium with Orthophthaldialdehyde (OPA)-derivatization reactions, such as OPA-sulphite reagent [5] or more recently OPA- tioglycolate [10] and OPA-NAC [4] by forming fluorescent derivates. Background fluorescence from naturally occurring organic compounds may not be negligible in some fresh waters and then the results obtained by the methods could be biased [5]. Chemiluminescence procedures have also been developed for ammonium determination [11-12], due to its high sensitivity, rapidity, simplicity and feasibility. The reagent mainly used is luminol combined with hypochlorite and the procedures are systems of flow injection analysis (FIA), not being described its application by HPLC in order to improve selectivity. Despite the known reaction between nitrogen compounds and dansyl chloride reagent (Dns-Cl), the reaction has not been applied to the quantitative determination of ammonium ions with fluorimetric nor postcolumn chemiluminescence generation ( with reagents such Bis-(2,4,6-trichlorophenyl)oxalate, TCPO) detection systems. However, dansylation has been widely used for the determination of analytes such as biogenic amines in plants [13] as low-molecular-mass amines applied to water samples [14-16],
- 215. 191 phenols in surface waters [17], phenoxyl-type N-methylcarbamate pesticide residues in fruit juices [18] and amphetamines in urine [19-20]. Based on our experience in the derivatization of amine compound with Dns-Cl and TCPO post-column chemiluminescence generation [16, 19-20], in this work, a fast, sensitive and selective method, has been developed for the screening and quantification of ammonium in water samples at low concentration level. The solution derivatization of ammonium in the sample with dansyl chloride reagent was studied, followed by HPLC in order to separate ammonium signal from other nitrogen-compounds response. Optimization of the separation on a C18 analytical column and postcolumn mixing with TCPO/H2O2 was carried out. A screening procedure for aliphatic amines and ammonium in water samples of different nature has been developed. Ammonium determination was carried out in different kind of samples. Moreover, the method was applied to several samples after applying Kjeldahl treatment in order to obtain values of Total Kjeldahl Nitrogen. EXPERIMENTAL Apparatus The chromatographic system used consisted of a quaternary pump (Hewlett Packard Series 1050) equipped with a high-pressure six-port valve (Rheodyne model 7000). The volume of sample loop was 20 µl. The post column instrument (PCX 5100), from Pickering Laboratories (Mountain View, CA, USA), consisted of two isocratic pumps. The chemiluminescence detector was a Jasco CL-1525 (Ishika-cho, Hachioji-shi, Tokio, Japan) which was coupled in series and linked to a data system Borwin-chromatography software and the interface Hercule lite (JMBS, France) used for data acquisition and storage. The model CL-1525 detector is equipped with JASCO´s uniquely designed coiled PTFE tube flow cell placed inmediatelly next to a highly sensitive end-on photomultiplier. All the assays were carried out at room temperature. For sample Kjeldahl treatment, the Digestion Unit was a Tecator Digestion system 6, 1007 Digestor (Höganäs, Sweden), and the Distillation Unit was a Büchi 323 Distillation Unit (Switzerland). Reagents All the reagents were of analytical grade. Stock standard solutions of ammonium were prepared by dissolving ammonium chloride (Probus, Barcelona, Spain) in nanopure water (1000 mg/L). Acetonitrile (J.T.Baker, Deventer, Holland), Tetrahydrofuran (Fluka, Steinheim, Switzerland), Methanol and Acetone (Scharlau, Barcelona, Spain) were of HPLC grade. Methylamine (MA), Ethylamine (EA), N-Propylamine (N-Pra), Butylamine (BA), Dimehylamine (DMA), Diethylamine (DEA), Pentylamine (PeA), Hexylamine (HA) and Dansyl Chloride (Dns-Cl) were obtained from Sigma (St.Louis, MO). TCPO, bis (2, 4, 6-trichlorophenyl oxalate), tetrahydrofuran and Imidazole (99%) from Fluka
- 216. 192 (Steinheim, Switzerland), and H2O2 (30%), sodium carbonate and sodium hydroxide from Merck (Darmstadt, Germany) were also used. Sodium thiosulphite and potassium sulphate from Prolabo (Fontenay, France), sodium hydroxide, sulphuric acid and sodium carbonate from Merck (Darmstadt, Germany), and red mercury oxide from Panreac (Barcelona, Spain) were used for sample Kjeldahl treatment. Columns and Mobile -Phases A LichroCART RP 18 (125x4 mm i.d., 5 µm particle φ) (Merck, Darmstadt, Germany) column was used for separation of ammonium derivative from reagent excess and from other nitrogen-compound derivatives. An acetonitrile:imidazole solution (1 mM, pH=7.0) (50:50 v/v) mixture in gradient elution mode was used as eluent at flow rate of 1 ml/min. The mobile phase gradient used was the following: gradient elution mode 1: acetonitrile:imidazole 50:50 at zero time, 70:30 at 6 min, and 50:50 at 7 min. When amine standards were studied, the gradient elution mode was extended in order to assure the elution of all amine derivatives following the gradient elution 2, established in our previous studies [15]: acetonitrile:imidazole 50:50 at zero time, 90:10 at 9 min, and 50:50 at 10 min. All the solvents were filtered with a 0.45 µm nylon membrane (Teknokroma, Barcelona, Spain) and degassed with Helium before use. Preparation of solutions Standard solutions of ammonium were prepared by dissolving the pure compounds in water (1000 µg/ml). Working ammonium solutions were prepared by diluting the standard solutions in water. Dns-Cl 5 mM solution was prepared by dissolving the pure compound in acetonitrile. All solutions were stored in the dark at 4 ºC. Solution derivatization of ammonium Ammonium was derivatized according to the method described by Marce et al. [13] with some modifications. To 1.55 ml of ammonium standard, 0.05 ml of 200 mM carbonate buffer (pH 9), 0.28 ml of acetonitrile and 0.12 ml of 5 mM dansyl chloride (in acetonitrile) were added successively, and the mixture was incubated at 70 ºC for 10 min. An aliquot (20 µl) of the solution was injected into the HPLC system.
- 217. 193 Post-column generation of chemiluminescence H2O2 (11 mM, prepared in acetonitrile) and TCPO (2.5 mM, prepared in acetonitrile:tetrahydrofuran (75:25)) were used as post column reagents. The post column system was running continuously during all runs at a flow rate of 0.35 ml/min, with TCPO (pump 2) and H2O2 (pump 3) (see the assembly on Figure 1). The dansyl derivatives, previously separated in the analytical column, were transferred to the post column system, where the intermediate generated from the TCPO and H2O2, served to excite the derivative so that it could undergo chemiluminescence emission. The distance between the reagents confluence and the detector was a tube of 5 cm (i.d. 0.508 mm). This was the minimum possible distance in the assembly. FIGURE 1. Assembly used for separation of dansylated ammonium and on-line post-column chemiluminescence detection. Selectivity Several aliphatic amines were derivatized following the same solution derivatization conditions as for ammonium determination but using gradient elution mode 2. Standards of 0.1 and 0.3 mg/l of MA and DMA, 0.2 and 0.6 mg/l of EA, 0.3 and 0.9 mg/l of N-PrA, BA, PeA and HA, and 0.5 and 1.5 of DEA were analysed. Analysis of real water samples The method was tested in real water samples of different origin and with unknown ammonium concentration. The environmental water samples were named as Post-column system Analytical column Acetonitrile : Imidazol buffer pH 7 Gradient elution mode 1 ml/min Pump 1 Hydrogen peroxide 0.011 M, 0.35 ml/min Pump 3 TCPO 2.5 mM 0.35 ml/min Pump 2 Chemiluminometer WW Sample
- 218. 194 bottled water (S1), tap water (S2), fountain water (S3), irrigation ditch water (S4)(S5) lake water (S6), sea water (S7) (S8) (S9), residual water from a factory (S10). Water samples were collected, filtered through 0.45 µm and acidified to pH 2 with HCl. Previously to the analysis, the samples were alkalised with NaOH to pH 6-7. Depending on the ammonium concentration in the sample, sample volumes ranged between 0.15 to 1.5 ml were taken. For bottled, tap, irrigation ditch and sea water 1.5 ml samples were processed. Lower volumes were taken for fountain (0.643 ml) and lake water samples (0.188 ml). For residual water sample 0.12 ml of 1:10 sample dilution was employed. The samples were also spiked with known amounts of the stock standard solutions of ammonium to give added concentrations of 0.15 and 0.3 mg/L. In all cases the volume of sample or sample plus spike was completed if necessary to 1.55 mL with deionized water. These volumes were processed according to the procedure described above for standard solutions. Kjeldahl treatment Kjeldahl treatment was applied to 100 ml of fountain water (S3), irrigation ditch water (S4), lake water (S6) and sea water (S9). For the application of Kjeldahl treatment [21], 6.7 g of potassium sulphate, 0.52 g of red mercury oxide and 10 ml of concentrated sulphuric acid, were added to 100 ml of sample in a digestion tube. Then, the tubes were placed on Kjeldahl digestor, which was previously heated at 370ºC. Heating was running until white fumes appeared and then, digestion continued for 30 minutes more. After that, digestion residue was cooled and diluted up to 100 mL with nanopure water. Distillation was performed by adding 35 ml of distillation reagent (25 g of sodium thiosulphate and 500 g of sodium hydroxide diluted up to 1L with nanopure water). Distilled ammonia was picked up on 25 ml of 0.04 N sulphuric acid until 80 mL. This solution was adjusted to pH 6-7 and diluted up to 100 mL. 1.55 mL of the resulted solution were processed according to the procedure described above for standard solutions. RESULTS AND DISCUSSION Optimization of precolumn ammonium dansylation Ammonium was derivatized according to the conditions established by Marcé et al.[13] for polyamines, with some modifications. For establishing the optimal conditions the dansylated derivatives were injected in the HPLC systems and the post column chemiluminescence generation with TCPO was performed according to the procedure described by our research group for aliphatic amines [16] (see experimental sectionand Figure 1) in order to register the chromatograms. The dansylated ammonium eluted at 2.4 min (Figure 2).
- 219. 195 FIGURE 2. Chromatograms corresponding to a blank reagent and standard solutions of ammonium of 0.19 mg/L. The chromatogram corresponding to the blank solution presented a peak at the same retention time than ammonium peak which was dependent on the amount of solvent (acetonitrile 1, Scharlau) used in the reagent solution. Different gradient elution modes were assayed in order to separate the two peaks, and also a precolumn filled with CN, C18, SCX or SAX packing, was inserted by means of a switching valve, but no separation was obtained, so it was deduced that the blank peak was due to pollution of organic solvent with ammonium. Therefore, other solvents were tested: acetone, acetonitrile 2 (J.T.Baker), tetrahydrofuran (THF), and methanol. The reagent mixture contained 0.9 mL of solvent (0.72 mL plus 0.18 mL of the reagent solution of Dns-Cl 5 mM), 0.2 mL of 100 mM carbonate buffer (pH 9) and 1 mL of nanopure water or ammonium standard. Although with solvents like methanol or THF no significant peak appeared for the blank at ammonium retention time, they were not appropriated due to in methanol the post-column reaction efficiency decreased and in THF the dansylation reaction did not take place. Acetone presented similar efficiency to acetonitrile 1, but the blank peak was also significative. In acetonitrile 2 (J.T.Baker), no significative peak appeared for blank register, and the signal for ammonium was good. According to these results, acetonitrile from J.T Baker solvent was selected for further experiments. Then, several volumes of the selected solvent were assayed. Figure 3 shows the influence of solvent volume on the blank signal. The presence of 0.4 mL of acetonitrile in the reaction mixture was selected as optimum due to provided the lowest blank signal without losing in ammonium response. 0 1 2 3 4 Time (min) ChemiluminescenceSignal NH4+ Blank Standard
- 220. 196 FIGURE 3. Influence of solvent volume, employed in the reaction derivatization with dansyl chloride (Conditions: DnsCl 0.45 mM), and reagent concentration (Dns-Cl) (Conditions: Acetonitrile Volume 0.4 mL) on the blank signal. The reagent concentration (Dns-Cl) in the mixture reaction was also studied in the range 0.15-0.75 mM maintaining constant the acetonitrile volume at 0.4 mL. 0.3 mM was chosen as optimum reagent concentration because provided a low blank signal (Figure 3) without losing in ammonium response. According to these results the optimal conditions were those shown in Table 1. In this table are compared the conditions established by Marcé et al.[13] and those optimized in this paper, as can be seen the original procedure has been simplified in order to make it quicker. TABLE 1. Comparison of the derivatization procedure for solution derivatization according to Marcé et al and for that proposed in this paper. Marcé et al. Procedure[13] Procedure optimized in this work 200 ì L sample/standard + 40 ìL internal standard + 200 ìL saturated sodium carbonate + 400 ì L DnsCl 5 mg/ml in acetone 1.55 mL sample/standard + 50 ìL carbonate buffer 0.2 M pH 9 + 400 ìL DnsCl 1.5 mM in acetonitrile Incubate in the dark overnight at room temperature or for 10 min at 70ºC Incubate 10 minutes at 70ºC Add 100 ìL of proline solution (100 mg/ml) and incubate in the dark for 30 minutes Inject 20 ì L onto the HPLC system Extract dansylated polyamines in 500 ìL of toluene and mix for 30 seconds Remove 400 ìL of organic phase, dry, and redissolve with 800 ìL of acetonitrile Pass the sample through a 0.45 ìm pore size syringe filter Inject 20 ì L onto the HPLC system 0 10000 20000 30000 40000 50000 60000 70000 80000 0 0.2 0.4 0.6 0.8 1 Acetonitrile Volume (mL) or DnsCl Concentration (mM) BlankPeakHeigh(V) Variable Acetonitrile Volume Variable DnsCl concentration Optimum conditions
- 221. 197 Analytical performance characteristics Table 2 shows the calibration graph obtained in the best conditions. Good linearity was obtained between 0.027-0.75 mg/L of ammonium and the detection limit established as the concentration required to generate a signal-to-noise ratio of 3, was 8 µg/L. The chromatogram corresponding to detection limit appears in Figure 4. The achieved detection limit is far away from legislation limit established (0.5 mg/L for non digested samples, and 2 mg/l for samples treated by Kjeldahl method). TABLE 2. Equations of the calibration graphs obtained in several conditions. The analytical signal used was peak height, except (*) where area instead of height was employed. Condition Dns-Cl (mM) NaCl Calibration curve Y = (a ±± sa) + (b ±± sb)· C (n, r2 ,sy/x) Detection Limit (µg/L) Linear Interval (mg/L) [1] 0.15 Y = (12 ± 6) + (130 ± 10)· C (5, 0.9814,9) 60 0.28-1 [2] 0.3 Y = (2 ± 7) + (960 ± 30)· C (10, 0.9935, 16) Y* = (3 ± 90) + (12300 ± 400)· C (10, 0.9929, 205) 8 0.027-0.75 [3] 0.3 3.5 g/l Y = (14± 5) + (930 ± 30)· C (3, 0.9998, 3) 8 0.027-0.75 [4] 0.3 35 g/l Y =(19 ± 14) + (980 ± 70)· C (3, 0.9942, 16) 8 0.027-0.75 0 2 4 6 8 10 ChemiluminescenceSignal Time (min) NH4+ R MA EA N-PRA + DMA BA DEA PEA HA R 1 2 3 LD NH4+ FIGURE 4. Chromatograms corresponding to 1: blank, 2: ammonium standard of 8 µg/L and 3: mixture of amines: Methylamine (MA) 0.1 mg/L, Ethylamine (EA) 0.2 mg/L, N- Propylamine (N-PrA) 0.3 mg/L, Dimethylamine (DMA) 0.3 mg/L, Butylamine (BA) 0.3 mg/L, Diethylamine (DEA) 0.5 mg/L, Hexylamine (HA) 0.3 mg/L and ammonium (NH4+) 1.5 mg/L.
- 222. 198 Table 2 also reports the calibration graphs obtained by using a smaller reagent concentration and the optimized reagent concentration in presence of different salinities. The smaller concentration gave only 13 % of the sensibility provided by the optimized reagent concentration. The slope values obtained for the salinities assayed were statistically similar to that obtained in absence of NaCl at 95 % confidence level. Then, the method is not influenced by the salinity. The study of interday and intraday precision at different concentrations levels can be seen in Table 3. The method provided good precision for interday and intraday measures independently of the concentration, with relative standard desviation (%RSD) between 2.6-7.2 % and 4-8.6 % respectively. So, repeatibility and reproducibility of the method resulted satisfactory. The accuracy of the method was also tested analysing several standards of known concentration, and as can be seen in Table 3, the results were suitable with relative errors (%Er) between 5-9.8%. TABLE 3. Interday and intraday precision (%RSD), and Accuracy (%Relative error, %Er), provided by the method for different concentrations of ammonium. n, number of standards. Interday precision Intraday precision Accuracy NH4 + (mg/l) %RSD (n) NH4 + (mg/l) %RSD (n) NH4 + (mg/l) %Er (n) 0.11 6.2 (5) 0.028 5.8 (4) 0.15 7.2 (4) 0.165 8.6 (5) 0.055 5 (4) 0.2 4.0 (3) 0.165 5.9 (3) 0.3 2.6 (4) 0.5 8.4 (3) 0.193 9.8 (3) In order to study the selectivity of the method, and knowing the reactivity of amino-compounds towards Dns-Cl reagent, amine interference was studied. Several aliphatic amines, such as MA, EA DMA, N-PrA, BA, DEA, PeA and HA were tested. Indeed, all these amine reacted with Dns-Cl, but their derivatives showed retention times different of the ammonium dansylated derivative (see Figure 4). It was studied the possibility of reagent consumption in presence of the different amines and ammonium at concentration level of 0.1 mg/L. The ammonium recovery in a solution containing the amines was 89.3%. Then, no significant differences were observed when the reaction took place in presence of all the amines tested. Analysis of real water samples The proposed method was applied to the analysis of several real water samples (see experimental section). As an illustrative example, in Figure 5, are shown the chromatograms in triplicate obtained for residual (A) and lake (B) water. Spiked samples with known amounts of analytes were used to validate the method accuracy. When Standard Additions Method (MOSA) was applied, Natural (S1), tap (S2), fountain (S3), irrigation ditch 2(S5), lake (S6) and residual (S10) waters
- 223. 199 presented similar slopes to that obtained by the calibration graph with standards (see Table 2) at 95 % confidence level as t tests [22] given in Table 4 show, therefore no matrix effect was found. For sample S5 (irrigation ditch 1) both slopes, MOSA and ammonium standard equation, were similar at 98 % confidence level. For the sea waters only S7 provided similar slopes at 99 % confidence level, while S8 and S9 gave matrix effect as can be seen in Table 4. When matrix effect is present, ammonium concentration must be calculated by applying MOSA. The MOSA equations (ordinate: a(sa), slope:b(sb), r2 , n, sy x) for S8 and S9 samples were : 57(5), 490(30), 0.9976, 4, 8.8 and 55(5), 320(30), 0.9707, 5, 8.6, respectively. The MOSA slopes were smaller than that corresponding to the ammonium standard calibration graph (Table 2). TABLE 4. t test for the comparison of the slopes of the ammonium standard calibration graph and of the MOSA equations for all samples tested. The tabulated values correspond to 95 % confidence level except * (98 %) and **(99 %). Sample Calculated F Tabulated F Conclusión 1 Calculated t Tabulated t Conclusión 2 S1-Bottled 3.988 17.443 Homogenous variances 1.086 2.7767 Similar slopes S2-Tap 6.150 799.5 Homogenous variances 2.940 3.1824 Similar slopes S3-Fountain 3.205 864.16 Homogenous variances 0.614 2.7764 Similar slopes S4-Irrigation ditch 1 1.606 864.16 Homogenous variances 3.540 2.7764 3.7469* Similar slopes S5-Irrigation ditch 2 92.003 17.443 Non homogenous variances 2.024 12.603 Similar slopes S6-Lake 44.31 17.443 Non homogenous variances 1.264 12.496 Similar slopes S7-Marine 1 2.23 799.5 Homogenous variances 3.894 3.1824 5.84** Similar slopes S8-Marine 2 5.36 17.443 Homogenous variances 5.960 2.7764 4.6041** Matriz effect S9-Marine 3 5.421 17.443 Homogenous variances 8.137 2.7764 4.6041** Matriz effect S3- Kjeldahl 1.659 647.79 Homogenous variances 0.071 4.3027 Similar slopes S4- Kjeldahl 2.378 16.044 Homogenous variances 0.057 2.5707 Similar slopes S6- Kjeldahl 2.726 647.79 Homogenous variances 2.311 4.3027 Similar slopes S9- Kjeldahl 3.409 864.16 Homogenous variances 0.619 2.7764 Similar slopes S10- Kjeldahl 1.571 799.5 Homogenous variances 1.179 3.1824 Similar slopes
- 224. 200 FIGURE 5. Chromatograms corresponding to water samples (in triplicate): A) Residual water (0.12 mL of 1:10 diluted sample to 1.55 mL). B) Lake water (0.188 mL of sample to 1.55 mL). The found spiked concentrations calculated employing the ammonium standard calibration graph are shown in Table 5. Relative errors in concentration were below 10.7 % in all cases except sample S7, which presented MOSA slope similar to that provided by ammonium standards at 99 % confidence level. Chromatograms for the fountain water fortified with 0, 0.15 and 0.3 mg/L of ammonium are shown in Figure 6. TABLE 5. Found ammonium concentration (mg/l) in the real samples spiked at different concentration levels, assayed directly or after Kjeldahl treatment. In brackets, %Error relative found for every sample at every concentration level. Free ammonium After Kjeldahl treatment Added NH4 + 0.15 mg/l Added NH4 + 0.3 mg/l Added NH4 + 0.15 mg/l Added NH4 + 0.3 mg/l Water Sample Found concentration (%Error) Found concentration (%Error) S1-Natural 0.155 (3.3%) 0.268 (10.7%) S2-Tap 0.141 (6%) S3-Fountain 0.135 (9.3%) 0.299 (0.3%) 0.138 (8%) 0.299 (0.3%) S4-Irrigation ditch1 0.145 (3.3%) 0.268 (10.7%) 0.141 (6%) 0.278 (7.3%) S5-Irrigation ditch2 0.135 (10%) 0.271 (9.7%) S6-Lake 0.145 (3.3%) 0.283 (5.7%) 0.169 (12.7%) 0.327 (9%) S7-Marine1 0.189 (26 %) 0.354 (18%) S9-Marine3 0.147 (2%) 0.291 (3%) S10-Residual 0.149 (0.3%) 0.304 (1.3%) 0.148 (1.3%) 0.320 (6.7%) 0 1 2 3 4 Time (min) ChemiluminescenceSignal NH4+ NH4+ A) B)
- 225. 201 FIGURE 6. Typical chromatograms for the fountain water (0.643 mL of sample to 1.55 mL) fortified with 0, 0.15 and 0.3 mg/L of ammonium. n in the last column is the number of solutions used for applying MOSA. In Table 6 is described the screening response of the proposed method to all samples assayed taking as reference the concentration limit established by legislation. As can be seen, ammonium was found in S3, S6, S7, S8, S9 and S10, but only residual water (S10) and Marine water (S7) exceed the limit established by legislation (0.5 mg/L). Table 6 also shows the ammonium content found by using the calibration graph of ammonium (see Table 2) and MOSA method providing similar results. Fountain and residual waters were also analysed by using OPA-NAC method [4]. The results were: 0.37(s=0.02, n=3) and 32(s=0.4, n=3), respectively. Both values can be considered statistically equal (at a confidence level of 95 % ) to those obtained by the proposed method. The quantified ammonium in residual water should be observed with reserve due to the high dilution carried out in the sample for both chemiluminescence and fluorescence methods. A representative group of these samples were treated to evaluate Total Kjeldahl Nitrogen (TKN). As t tests given in Table 4 indicate the slope of the MOSA method is similar to that corresponding to the standard calibration graph of ammonium. Therefore neither of the samples presented matrix effect even sea water and the ammonium concentration in the samples was obtained directly from the calibration graph with standards of ammonium (Table 5). The samples were also spiked with known amounts of ammonium in order to validate the accuracy and the found ammonium concentrations are shown in Table 4. As can be seen, low relative errors between 0.3 and 10.7% were achieved. 0 2 4 6 ChemiluminescenceSignal Time (min) S3 S3 + 0.15 mg/l NH4+ S3 + 0.3 mg/l NH4+ NH4+ NH4+ NH4+
- 226. 202 Ammonium concentration was higher after Kjeldahl treatment in all samples assayed. These results are shown in Table 6. As can be seen in this Table, fountain, residual and lake water, gave a positive screening response, being its ammonium concentration higher than limit established by legislation (2 mg/L). TABLE 6. Screening response and ammonium concentration deduced on different samples assayed.* Estimative values due to the high dilution of the sample carried out. Sample Legislation limit (mg/l) Screening response Ammonium Conc (C ±± s) (mg/l) (n=3) Standard Calibration Ammonium Conc (C ±± s) (mg/l) (n) MOSA S1-Natural 0.5 No - - S2-Tap 0.5 No - - S3-Fountain 0.5 No (0.367 ± 0.012) 0.347 (n=5) S4-Irrigation ditch1 0.5 No - - S5-Irrigation ditch2 0.5 No - - S6-Lake 0.5 No (0.352 ± 0.014) 0.378 (n=5) S7-Marine1 0.5 Yes (3.8 ± 0.1) 3.108 (n=5) S8-Marine2 0.5 No Matrix effect 0.098 (n=5) S9-Marine3 0.5 No Matrix effect 0.147 (n=5) S10-Residual 0.5 Yes (30 ± 4) 30.69 (n=4) S3 Kjeldahl 2 Yes (2.28 ± 0.12) 2.34 (n=3) S4 Kjeldahl 2 No (0.56 ± 0.10) 0.595 (n=4) S6 Kjeldahl 2 Yes (3.05 ± 0.13) 2.71 (n=3) S9 Kjeldahl 2 No (0.81 ± 0.08) 0.885 (n=5) S10 Kjeldahl 2 Yes (169 ± 18) 152.6 (n=4) CONCLUSIONS This paper studied the chemiluminescence detection of ammonium in order to improve selectivity and sensitivity. The procedure used solution dansylation. The dansylated derivative was chromatographed and mixed with TCPO/H2O2 in order to generate chemiluminescence. The quantification of ammonium at low levels (0.027-0.75 mg/l) is possible with satisfactory accuracy (errors below 9.8%) and precision (%RSD between 2-8.6%). The proposed method is selective in front of amine presence in the sample and it can be applied for the screening and quantification of ammonium in water samples, being detectable even concentrations of 8 µg/L, which are far away from limit established by legislation (0.5 mg/l for non-digested water samples, and 2 mg/l for samples treated by Kjeldahl procedure). Ammonium can be quantified from standard calibration curve for almost all kind of waters tested: bottled, tap, lake, fountain, irrigation and residual. However, two of the
- 227. 203 three assayed marine water samples required the application of standard additions method for the quantification of ammonium due to the presence of matrix effect but not when the Kjeldahl treatment is applied. Acknowledgements The authors are grateful to the Ministerio de Ciencia y Tecnología of Spain for financial support received for the realization of Project BQU2003-06138 . Susana Meseguer Lloret expresses her gratitude to Ministerio de Educación y Cultura (Spain) for the predoctoral grant. Bibliographic References [1] http:/www.atsdr.cdc.gov/es/toxfaqs/es_tfacts33.pdf [2] European Directive 98/83/CE. [3] Díaz-Santos S.A. Métodos Normalizados para el Análisis de Aguas Potables y Residuales. Editorial Apha-Awwe-WPCF. 17 Ed. (1992). [4] S.Meseguer Lloret, C.Molins Legua, P.Campins Falcó, Intern.J.Environ.Anal.Chem., 82 (2002) 475-489. [5] A. Aminot, R.Kerouel, D.Birot, Wat.Res. 35-7 (2001) 1777-1785 [6] J.P. Hart, A.K. Abass, D.C. Cowell, A. Chappel, Electroanalysis, 11(1999) 406-411 [7] J. Schwarz, H. Kaden, G. Pausch, Fresenius J. Anal. Chem.., 367(2000) 396-398. [8] M-Mori, K.Tanaka, M.I.H.Helaleh, Q.Xu, M.Ikedo, Y.Ogura, S.Sato, W.Hu, K.Hasebe, J.Chromatography A, 997 (2003) 191-197. [9] D.H.Thomas, M.Rey, P.E.Jackson, J.Chromatography A, 956 (2003) 181-186. [10] H.Mana and V.Spohn, Fresenius J Anal Chem, 366, 825-829 (2000). [11] J.Li, P.K.Dasgupta, Anal.Chim.Acta., 398 (1) (1999) 33-39 [12] W.Qin, Z.J.Zhang, B.X.Li, Y.Y.Peng, Talanta 48 (1999) 225-229. [13] M.Marcé, D.S.Brown, T.Capell, X.Figueras, A.F.Tiburcio, J. Chromatog. 666 (1995) 329-335. [14] M.Cobo, M.Silva, J. Chromatog. A, 848 (1999) 105-115. [15] S. Meseguer Lloret, C. Molins Legua and P. Campins Falco, J. Chromatog. A, 978 (2002) 59-69. [16] S. Meseguer Lloret, C. Molins Legua, J. Verdú Andrés, P. Campins Falco, J. Chromatog. A, in revision. [17] P.J.M.Kwakman, D.A.Kamminga, U.A.Th.Brinkman, J. Chromatog. 553 (1991) 345-356. [18] E.Orejuela, M.Silva, J. Chromatog. A, 1007 (2003) 197-201. [19] C.Molins-Legua, P.Campins-Falco, A.Sevillano-Cabeza. Anal. Chim, Acta. 378 (1999) 83-93. [20] C.Molins-Legua, P.Campins-Falco, A.Sevillano-Cabeza, Analyst. 123 (1998) 2871- 2876. [21] Application Note ASN 52/85. Tecator Manual Digestion system 1007 (1985). [22] D.L. Massart, B.G.M. Vandeginste, L.M.C. Buydens, S. De Jong, P.J. Lewi y J. Smeyers-Verbeke. Handbook of Chemometrics and Qualimetrics: Part A, Elsevier, Amsterdam 1997.
- 228. 204
- 229. 205 Apéndice 6. TALANTA, REVISED PAPER FI AUTOMATIC METHOD FOR THE DETERMINATION OF COPPER(II) BASED ON COPROPORPHYRIN I - Cu(II) / TCPO / H2O2 CHEMILUMINESCENCE REACTION FOR THE SCREENING OF WATERS S. Meseguer-Lloreta , P. Campíns-Falcóa , S. Cardenasb , M. Gallegob and M. Valcárcelb * a Departamento de Química Analítica, Universidad de Valencia, Spain b Departamento de Química Analítica, Campus de Rabanales, Universidad de Córdoba, Spain Abstract In this paper, an automatic method for the screening of water samples containing Cu(II) was proposed based on peryoxalate chemiluminescence reaction, using Coproporphirin I as fluorophor compound to provide selectivity, and a simple FI chemiluminescence detector. FI system conditions were chosen in order to distinguish samples over or under legislation limit established (50 µg/l), with high reliability. The detection limit found was 9 µg/l and the linear dynamic range was from 15-125 µg/l of Cu(II). Repeatibility and reproducibility studies gave good precision and accuracy with %recovery near 100%. In that conditions, the method resulted selective and only Fe(II), Fe(III) and Pb(II) could interfere, but at concentration level higher than their normal concentration in waters. The proposed method was found to be reliable for screening purposes and it was successfully applied to a variety of real water samples. * Correspondence author. Tel./fax: + 34 957 218 616. E-mail address: qa1meobj@uco.es
- 230. 206 1. Introduction The chemiluminescence technique provides methods for trace analysis that are attractive because of their high sensitivity and fast kinetics. This technique, combined with FI systems provides good accuracy and precision. Peryoxalate chemiluminescence reaction (TCPO/H2O2 system), has been extensively used in recent years for liquid chromatography of fatty acids [1], synthesis, characterisation and analytical evaluation of oxamide reagents [2], determination of amphetamines in urine samples [3], inmunoassays [4], detection of proteins separated by capillary zone electrophoresis [5], resolution of carboxylic acids by HPLC [6], determination of cholesterol [7], detection of retinoids using normal-phase chromatography [8] and the study of emission of spectroscopic properties of water soluble porphyrins with d- and f- electron metals [9, 10]. The use of porphyrins can be remarked by their high selectivity to metal ions [10]. Metal ions are usually introduced into water cycle by industrial dumping and many of them present toxic effects for human life; therefore, their maximum tolerated concentration have been fixed by legislation, being for copper(II) 50 µg/l [11]. In order to control these values in real water samples, fast automatic systems are required; these type of systems are called screening systems [12, 13], and provide a binary yes/no response that let to distinguish samples containing Cu(II) over or under legislation level with good reliability, fast speed, versatility and low cost. Igarashi et al. [10] proposed an static method for Copper (II) determination based on coproporphyrin I-Cu(II)/TCPO/H2O2 chemiluminescence reaction. Based on their studies, we are proposing in this paper an automatic FI method for the screening of Cu(II) in water samples. 2. Experimental 2.1 Apparatus Flow injection chemiluminescence measures were carried out with a FireFly Chemiluminescence Detector (Global FIA Inc. P.O. Box 480, Fox Island, WA 98333) designed specially for use in flow through systems. Light generated in a liquid core waveguide is internally reflected and transmitted to a small but powerful photo multiplier. FI registers were monitored by an Hercule Lite-Chromatography interface connected on line to a personal computer. The flow system consisted of two Gilson Minipuls 3 peristaltic pump furnished with poly(vinyl chloride) pumping tubes, two Rheodyne (Cotati, CA) 5041 injection valves, PTFE tubing of 0.5 mm i.d., and a water bath provided by a heater magnetic stirrer (Raypa, Spain).
- 231. 207 Static chemiluminescence measures were registered with a Jasco FP-750 (Tokio, Japan) fluorescence spectrophotometer. For the kinetics measure, the light emission was monitored at 622 nm. The chemiluminescence was measured with a 1 cm path length quartz cell and the last reagent (H2O2) was injected using a Hamilton Digital syringe (Nevada, USA). 2.2. Reagents and standard solutions All chemical reagents were of analytical grade or better. Copper (II) sulphate pentahydrated, hydrogen peroxide 30%, nitric acid (Trace pur) 69% and sodium carbonate were purchased from Merck (Germany); coproporphyrin I dihydrochloride was from Aldrich Chemistry (Germany); Bis(2,4,6.trichlorophenyl) oxalate (TCPO) (Fluka Chemika, Switzerland) was prepared in tetrahydrofuran (Scharlau, Barcelona, Spain) : acetonitrile (J.T.Baker, Holland). Hexadecyltrimethylammonium chloride 25% (CTAC) (Fluka Chemika, Switzerland) was prepared in 0.1 M pH 7.4 sodium tetraborate decahydrated (Panreac, Barcelona, Spain); ammonium acetate was also from Panreac (Barcelona, Spain). Metal interference solutions were prepared from nitrate or chloride salt (Probus, Spain). 2.3. Sample preparation 5 ml (for Sample 1, 2, 3, 4), 9 ml (for Sample 5) or 3.65 ml (for Sample 6) of water samples were added to 0.25 mL of borax buffer 0.2 M pH 7.4 and 0.1 ml of 10-4 M coproporphyrin I and diluted up to 10 ml. Sample 1 was a synthetic natural water sample, prepared carrying 26.7 mg/l of Ca(II), 106.5 mg/l of bicarbonate, 4.3 mg/l of Mg(II), 5 mg/l of Chloride, 12 mg/l of sulphate and 1 mg/l of fluoride. For Standard Reference Material SRM® 1640, 3.72 mL of SRM® containing 85.2 µg/l of Cu(II) (Reference value), were added to 0.1 ml of 10-4 M coproporphyrin I and 1.9 ml of ammonium acetate 1 M. The pH was adjusted to 5.35 and the mixture was diluted up to 10 ml. Water samples were measured directly and fortifying sample with 31.77 µg/l of Cu(II) for S1, S2, S3, S4 and SRM; and with 50 µg/l for S5 and S6. 2.4. Screening procedure In order to analyse copper (II) in waters, a FI automatic system was employed. It can be seen in Figure 1. Two peristaltic pumps were used. Pump 1 consisted of two carrier streams; in one of them, standards from 15 to 125 µg/l of copper (II), 10-6 M in coproporphyrin I at 2.2 ml/min, pass through a 400 µl reactor on a water bath at 100 ºC where flow was stopped for 5 min. After that, flow was started for 25 seconds in which sample was cooled passing through one reactor of 200 µl and mixed with the other
- 232. 208 CTAC/H2O2 stream (at 0.7 ml/min) in the second 200 µl coil to fill the 50 µl sample loop. Pump 2 consisted of two water carrier streams. One of them at 5 ml/min passes through sample loop(IV1), and the other at 2.9 ml/min passes through TCPO loop(IV2), previously filled by means of a syringe. Injection valve positions were activated manually. When injection valves were in position 1 (loading), only nanopure water was reaching the chemiluminescence detector. Both injection valves must be switched to position 2 (injection) at the same time, in order to allow both sample and reagents plugs reaching detector simultaneously. The height of the FI signal obtained must provide the binary (yes/no) response. FIGURE 1: FI system designed for the screening of water samples for copper (II) using a quenching chemiluminiscence determination. IV, injection valve; CLD, chemiluminiscence detector. 3. Results and discussion 3.1. Checking the FireFly Chemiluminescence Detector The detector was checked following the FI procedure described by L.A. Tortajada et al. [14] for Cr(III)/luminol/H2O2 reaction. Table 1 shows log-log calibration curves and detection limits obtained by this investigation group in recent publications [14, 15, 16] using a Hitachi F-4500 spectrofluorimeter and the same obtained with this new detector. IV1 IV2 CLD PUMP 1 (ml/min) PUMP 2 (ml/min) Standard/Sample (10 -6 M Porphyrin) CTAC 10 -2 M H2O2 0.7 M H2O H2O Water bath 100ºC 400 µl 200 µl 200 µl 50 µl 50 µl TCPO 0.7 2.2 5 2.9 L = 7.5 cm L = 12 cm
- 233. 209 TABLE 1: Comparison of sensitivity of detectors for Cr(III)/luminol/H2O2 chemiluminescence reaction. Reference (loga ±± sloga) (b ±± sb) Detection limit (µµg/l) Detector [14] (1.91 ± 0.03) (1.19 ± 0.03) 1.3 Hitachi F4500 [15] (1.97 ± 0.07) (1.25 ± 0.07) 1.2 Hitachi F4500 [16] (1.84 ± 0.03) (1.16 ± 0.03) 1.6 Hitachi F4500 This work (1.04 ± 0.06) (1.25 ± 0.06) 1.4 FireFly CLD 3.2. Optimization of the flow system Emission chemiluminescence spectrum was obtained for a blank solution following static conditions (1.12 µM coproporphyrin I, 0.0096 M CTAC, 1.795 mM TCPO and 0.743 M H2O2). In Figure 2a) it can be seen that there is an emission maximum at 622 nm. Reaction kinetics curve at 622 nm was also obtained. Figure 2b) shows that the kinetics of this reaction was very fast, it took place in about 10 seconds. FIGURE 2: a) Emission chemiluminescence spectrum for a blank. b) Reaction Kinetic curve. Conditions: 1.12· 10 -6 M coproporphyrin I, 0.0096M CTAC, 0.743 M H2O2, 1.795 mM TCPO. Optimization of the flow system was carried out with a blank solution using only pump 2 and injecting both coproporphyrin I 10-5 M/H2O2 variable/CTAC variable/borax 0.1M pH 7.4 (IV1, loop 160 µl) and variable TCPO (variable ratio MeCN:THF) (IV2, loop 200 µl) with a syringe (see Figure 1). Table 2 shows the variables optimized, the range studied on each case and the optimum variable selected. Firstly, the addition of CTAC was studied based on previous 0 20 40 60 80 100 0 50 100 Time (seconds) ChemiluminescenceSignal b) 0 0.2 0.4 0.6 0.8 1 400 500 600 700 λλ (nm) ChemiluminescenceSignal a) Maximum 622 nm
- 234. 210 studies [10]. Hydrogen peroxide and TCPO concentrations were varied according to recent studies of the chemiluminescence reaction [2, 10]. These two reagents must reach the detector separately in order to achieve the maximum sensitivity. TABLE 2: Optimization of the chemical and physical variables of the FI system. Variable Range studied Optimum condition CTAC conc 2· 10-5 – 9· 10-2 M 1· 10-2 M H2O2 conc 0.1 – 1 M 0.7 M TCPO conc 1 – 10 mM 5 mM MeCN:THF ratio 0%:100% – 100%:0% 75%:25% pH 4.7-10.3 7.4 Flow rate Pump 2 10 - 48 rpm 40 rpm Loop Volume 50 – 500 µl 50 µl Length from injection valve (IV) to CLD LIV1-CLD= 12 - 24 cm LIV2-CLD=7.5 - 15 cm LIV1-CLD= 12 cm LIV2-CLD=7.5 cm Length liquid core waveguide 17 – 50 cm 17 cm Coproporphyrin-Cu(II) Reaction time 1 – 10 min 5 min TCPO reagent was prepared in acetonitrile:tetrahydrofuran at different ratios from 0:100 to 100:0. Best results were obtained for 75% acetonitrile:25% tetrahydrofuran avoiding TCPO destruction and obtaining the best sensitivity. CTAC/H2O2 reagent pH was studied using different buffer solutions at 0.1 M concentration: pH 4.7 was studied in acetic/acetate buffer, pH 6.5 and 10.3 were studied with carbonate/bicarbonate buffer and pH 7.4 was studied with borax buffer. Best results were obtained at pH 7.4 with borax buffer. Concerning the flow conditions, flow rate in pump 2 was varied from 10 to 48 rpm, and 40 rpm was selected as optimum, providing 5 ml/min for coproporphyrin I stream and 2.9 ml/min for TCPO stream. Loop volume was also studied, and 50 µl were selected because signal was more reproducible and pressure problems were avoided. Due to the fast speed of the chemiluminescence reaction, another parameters were optimised: distance from injection valves to chemiluminescence detector and length of the liquid core waveguide. The length from injection valves (IV1 and IV2) to chemiluminescence detector (CLD) (see Figure 1) was selected as the minimum possible. This condition avoided reagent dispersion and improved sensitivity 10 times. The length of the liquid core waveguide was also selected as the minimum possible for the detector, and it improved the sensitivity about 2.5 times.
- 235. 211 With those changes, coproporphyrin I concentration could be reduced to 1· 10-6 M (lower coproporphyrin concentrations did not give reaction with Cu(II) with the established conditions). Then, Pump 1 was added to the FI system and copper (II) was introduced using the best conditions established for the optimisation of the Cu(II)- coproporphyrin reaction time in the water bath; it was studied from 1 to 10 min, and it was observed that quenching effect was better when reaction time increased, and 5 min were selected. 3.3. Sensitivity and selectivity of the method Calibration curves for Cu(II) were obtained in different conditions of Pump 1 flow rate and reaction time. Analytical figures of merit for the conditions assayed can be seen on Table 3. When Pump 1 flow rate was 0.7 ml/min for coproporphyrin carrier and 0.5 ml/min for CTAC/H2O2 carrier, calibration curves were obtained stopping flow for 5 min in the heating bath (t1) and 2 or 5 min in the second reactor of the flow system (t2) in order to cool the reaction mixture. Good reproducibility for the same conditions can be observed comparing the first and second calibration curve, being %Recoveryb of 97%. Increasing t2 from 2 min to 5 min, increased the sensitivity 1.65 times. TABLE 3: Cu(II) calibration curves and figures of merit in different conditions. (LI: linear interval; DL: detection limit). vpump1 (ml/min) (porph.carrier/ CTAC-H2O2 carrier) t1 (min) t2 (min) a ±± sa b ±± sb LI (µµg/l) DL (µµg/l) n, r2 , sy/x /b 0.7/0.5 5 2 4300 ± 40 -18.3 ± 0.7 0-125 17 12, 0.984, 5.38 0.7/0.5 5 2 4370 ± 110 -18.9 ± 1.3 0-125 17 8, 0.972, 8.96 0.7/0.5 5 5 6770 ± 30 -30.7 ± 0.4 0-125 10 5, 0.999, 1.32 2.2/0.7 4 0 3840 ± 20 -19.0 ± 1.0 0-32 17 4, 0.994, 1.22 2.2/0.7 5 0 4255 ± 18 -36.6 ± 1.0 0-32 8.7 4, 0.999, 0.58 2.2/0.7 5 0 5010 ± 50 -30.7 ± 0.8 0-100 10 10, 0.994, 2.83 When Pump 1 flow rate was fixed at 2.2 ml/min for coproporphyrin carrier and 0.7 ml/min for CTAC/H2O2 carrier, the flow was not halted in the second coil in order to eliminate a dispersion phase. Calibration curves were obtained for t1 = 4 or 5 min in the interval 0-32 µg/l. Results showed that sensitivity was increased near 2 times for t1= 5 min, obtaining the best detection limit at the level of 8.7 µg/l, level that was 5.8 times lower than limit established by legislation for Cu(II) in waters (50 µg/l). Calibration curve was also obtained with that conditions in the range from 0 to 100 µg/l which was the linear dynamic range because signal remained constant at higher Cu(II) concentrations. FI registers obtained can be seen on Figure 3. Limits of detection, which are shown in Table 3, were calculated as 3· sblank/b, being sblank = 105.96 (n=9).
- 236. 212 FIGURA 3: Cu(II) calibration curve FI registers. Repeatability and reproducibility studies were carried out for standards analysed in triplicate. Repeatability studies were obtained for a standard of 15.9 µg/l with a %RSD of 16%. Results for reproducibility studies were obtained for a standard of 17.3 µg/l of Cu(II), and %RSD was 12%. The recoveries obtained for repeatibility and reproducibility studies were 93 % and 109 %. These values are in accordance with the required accuracy in trace analysis. Results obtained with the Standard Reference Material SRM® 1640 analysed, demonstrated the accuracy of the method, giving a Copper (II) concentration found of (96 ± 14) µg/l (Reference value of SRM 1640: 85.2 µg/l). The percentage of recovery was (113 ± 17) % (n=3). In order to study the selectivity of the method, several metal ions were studied with this FI system. Na+ , Mg2+ , K+ , Ca2+ , Ag+ , Mn2+ , Ni2+ and Zn2+ only interfere at high 0.00 4.00 8.00 Time (min) 0.00 2000.00 4000.00 6000.00 ChemiluminescenceSignal 0 µg/l 31.8 µg/l 47.7 µg/l 79.4 µg/l 95.3 µg/l
- 237. 213 levels, between 5 and 500 mg/l, which are not usual in water samples. Al(III), Sn(II), Pb(II), Hg(II), Fe(II), Fe(III), Cd(II), Cr(III) and Co(II) were also studied. Their limit of tolerance (concentration at which signal was modified more than 10%) was lower, but this was over limit established by legislation. Only Pb(II), Fe(II) and Fe(III) had their limit of tolerance under limit established by legislation, but it was over its usual concentration in waters. Therefore, we concluded that the method was selective to Cu(II). These results can be observed on Table 4. Table 4: Interference study. Interference ion Legislation limit (µµg/l) Concentration in natural waters (µµg/l) Limit of tolerance (µµg/l) Al(III) 10 3000 Fe(III) 2000 500 2000 Fe(III) 2000 500 2000 Sn(II) 300 Pb(II) 50 1 30 Hg(II) 1 0.07 300 Cd(II) 5 0.03 30 Co(II) 0.05 30 Cr(III) 50 1 3000 3.4. Reliability of the screening method for Cu(II) determination The confidence level of the proposed sample screening system can be established on the basis of the percentage of false positives and false negatives through a simple chemometric study. The cut-off level was set at a concentration corresponding to two times the detection limit (2DL). A false positive corresponded to a water sample containing a Cu(II) concentration lower than the cut-off level but giving a positive response. A false negative is obtained when a water sample with an analyte concentration higher than that of the cut-off, provides a negative response in the detector. About 65 standards were measured at concentrations near detection limit: 0.5LD, 1LD, 1.5LD, 2LD, 3LD, 3.5LD µg/l. In Figure 4 can be seen that the percentage of false positive was 0% at 0.5DL, increasing at 9% for 1DL and 25% for 1.5DL. The percentage of false negatives was 6% for 3DL and 0% at 3.5DL. Considering that his value was far away from legislated value (50 µg/l), the proposed procedure can be used for the screening of Cu(II) in water samples.
- 238. 214 -50 -40 -30 -20 -10 0 10 20 30 40 50 4,3 8,7 13 17,3 26 30,3 %FP%FN µg/l Detection Limit Cut-off level 0.5DL 1DL 1.5DL 2DL 3DL 3.5DL FIGURE 4: Reliability of the screening method. Standard solutions (n=65) of Cu(II) at concentration near detection limit(DL). %FP=Percentage of false positives; %FN=Percentage of false negatives. 3.5. Application to real water samples The method was applied to the determination of Cu(II) in real water samples (see Table 5). A synthetic water sample (S1), three natural water samples (S2, S3, S4), tap water sample (S5) and a fountain water sample (S6) were analysed and only fountain water sample gave a positive response. The recoveries obtained for all samples demonstrated the accuracy of the method being between 83 and 119% of recovery. TABLE 5: Results obtained in the Cu(II) determination in real water samples. Sample Screening response Conc. Found (C ±± s) (µµg/l) Concentration added (µµg/l) %Rec ±± s S1, Synthetic sample No - 31.77 92 ± 16 (n=6) S2, natural water No - 31.77 102 ± 19 (n=3) S3, natural water No - 31.77 100 ± 13 (n=4) S4, natural water No - 31.77 117 ± 7 (n=4) S5, tap water No - 50 83 ± 11 (n=3) S6, fountain water Yes (61 ± 5) (n=8) 50 119 ± 12 (n=3)
- 239. 215 4. Conclusions The proposed method can be used as an automatic system for screening of Cu(II) in water samples providing good results in terms of sensitivity, repeatability, reproducibility, precision, accuracy and selectivity. It allows filtering sample set of study with good reliability, low cost, versatility, speed and minimization of operator errors, providing the analytical information of client interest. Acknowlegements The authors are grateful to the Ministerio de Ciencia y Tecnología for financial support. S.M.Ll. expresses her gratitude to Ministerio de Educación y Cultura (Spain) for the predoctoral grant. Financial support from the Spanish DGICyT (Grant BQU2001- 1815) is gratefully acknowledged. Bibliography [1] Y. Ohba, N. Kuroda, K. Nakashima. Anal. Chim. Acta, 465 (2002) 101-109. [2] N.W.Barnett, R.Bos, R.N.Evams, R.A.Russell. Anal. Chim. Acta, 403 (2000) 145- 154. [3] C.Molins-Legua, P.Campins-Falcó, A.Sevillano-Cabeza. Anal. Chim. Acta, 378 (1999) 83-93. [4] H.A.H.Rongen, R.M.W.Hoetelmans, A.Bult, W.P.Van Bennecom. J. Pharm. Biomed Analysis, 12 (1994) 433-462. [5] T.Hara, J.Yokogi, S.Okamura, S.Kato, Riichiro. J. Chromatogr. A, 652 (1993) 361- 367. [6] T.Toyo’oka, M.Ishibashi, T.Terao. J. Chromatogr. A, 627 (1992) 75-86. [7] J.H.Mike, T.J.Cleland. Anal. Chim. Acta, 259 (1992) 73-78. [8] P.D.Bryant, A.C.Capomacchia. J. Pharm. Biomed. Analysis, 9 (1991) 855-860. [9] K.Staninski, M.kaczmarek, S.Lis, M.Elbanowski. J. Solid State Chem., in Press, Corrected Proof. [10] S.Igarashi, T.Nagoshi, T.Kotake. Anal. Letters, 33 (2000), 3271- 3283. [11] Real Decreto 928/88 legislación española. [12] M. Valcárcel, S.Cárdenas, M.Gallego. Trends Anal. Chem., 18 (1999), 685-694. [13] M. Valcárcel, S.Cárdenas, M.Gallego. Trends Anal. Chem., 21 (2002), 251-258. [14]L.A.Tortajada-Genaro, P.Campins-Falcó, J.Verdú-Andrés, F.Bosch-Reig. Anal. Chim. Acta 450 (2001) 155-173. [15] S.Meseguer-Lloret, P.Campíns-Falcó, L.A.Tortajada-Genaro, F. Blasco-Gómez. Int. J. Environ. Anal. Chem., 83 (2003) 405-416. [16] Y.Moliner-Martínez, S. Meseguer-Lloret, L.A.Tortajada-Genaro, P.Campíns-Falcó. Talanta 60 (2003) 257-268.
- 240. 216
- 241. 217 Apéndice 7. S. Meseguer-Lloret, P. Campíns-Falcó, L. A. Tortajada-Genaro, F. Blasco Gómez. Departament de Química Analítica, Facultat de Química, Universitat de Valencia A guide to avoid method bias of Chromium (III, VI) chemiluminescence determination by luminol-hydrogen peroxide reaction. Application to water samples. Intern. J. Environ. Anal. Chem. 83 (5) (2002) 405-416. Enlace URL: http://www.tandf.co.uk/journals/titles
- 242. 218
- 243. 219 Apéndice 8. Y. Moliner-Martínez, S. Meseguer-Lloret, L. A. Tortajada-Genaro, P. Campíns-Falcó. Departament de Química Analítica, Facultat de Química, Universitat de Valencia. Influence of water sample storage protocols in chemiluminescence detection of trace elements. Talanta 60 (2003) 257-258. Enlace URL: http://www.sciencedirect.com
- 244. 220
- 245. 221 Apéndice 9. P. Campíns-Falcó, L. A. Tortajada-Genaro, S. Meseguer-Lloret, F. Bosh-Reig. Departament de Química Analítica, Facultat de Química, Universitat de Valencia. Multivariate calibration applied to simultaneous determination of cobalt and chromium. Anal. Bioanal. Chem. 374 (2002) 1223-1229. Enlace URL: http://springerling.metapress.com
- 246. 222
- 247. 223 Apéndice 10. UNPUBLISHED PAPER MODIFIED ROTH´S FLUORIMETRIC METHOD FOR WATER SAMPLES: AMMONIUM AND PRIMARY AMINE GROUPS DETERMINATION AND UTILITY IN TOTAL NITROGEN KJELDAHL. S. Meseguer Lloret, J. Verdú Andrés, C. Molins Legua and P. Campíns Falcó* Department of Analytical Chemistry, University of Valencia, Dr. Moliner 50, 46100- Burjassot, Valencia, Spain. e-mail address: pilar.campins @ uv.es. Tel.: +34-96-3983002; Fax: 34-96-3864436. Abstract A method for the simultaneous determination of primary amino groups and ammonium ion was proposed. The method is based in solution derivatization with o- Phthaldialdehyde / N-acetyl-cisteine (OPA/NAC) and fluorescence measurement of the formed isoindols. Analytical characteristics and the description of the developed procedure are provided. Calibration graph was obtained for ammonium (up to 1.44 mg/L of N) and for methylamine (up to 0.282 mg/L of N) as primary amino group model compound. Bivariate and multivariate calibration models were tested. The limits of detection were 0.07 mg/L of N and 0.004 mg/L of N for ammonium and amine, respectively. The procedure was first applied directly to standard solutions containing ammonium and amine and secondly to digested solutions by Kjeldahl. The results obtained allowed to establish if the best digestion conditions are achieved, in order to perform the total amine conversion into ammonium. This procedure has been also applied to real samples (irrigation ditch water, residual water and fountain water) and the concentration of primary amine groups and ammonium has been evaluated. The results obtained after Kjeldahl digestion has allowed to estimate the total Keldahl N contained in the samples. The samples were also analysed by Nessler method and similar results were obtained.
- 248. 224 INTRODUCTION Ammonium is a micronutrient in water systems which is an important link in the nitrogen cycle of aquatic ecosystems. However, its determination is still a delicate task, due to it is high susceptibility to contamination, and the classic methods does not appear to be well controlled in many laboratories [1]. Ammonium can be found in superficial, subterranean or marine waters at low concentrations, about 10 µg/L. In residual waters it can be found at high concentrations, about 30 mg/L due to ammonification and nitrate reduction processes. Often, it can be found together with short-chain aliphatic primary amines (mainly methylamine) because they are widely distributed in the environment. Aliphatic amines are used in several chemical and manufacturing industries and are also common components of biological systems as degradation products of organic material such as amino acids and proteins. Besides hygienic problems due to the stinging smell, these compounds may be hazardous to human health. In addition, they can react with certain nitrogen-containing compounds to form nitrosamines, which are potentially carcinogenic substances [2]. Consequently, there is an increasing interest in the determination of aliphatic amines in various aqueous matrices. The tolerable limits for amines are regulated as Kjeldahl Nitrogen (which include ammonium and organic nitrogen), being between 15 and 85 mg/L for residual waters and 1 mg/L for tap water. Liquid Chromatography combined [3] or not with derivatization [4], is generally used for the analysis of ammonium and aliphatic amines in aqueous media. Unbiased UV-vis or fluorimetric methods are not found in the literature, which could be used as screening sample methods in order to reduce costs and saving time in the environmental laboratory or in situ determinations. The Roth´s fluorimetric method [5-7] for the determination of primary amino compounds by use of a thiol and o-Phthaldialdehyde (OPA) as derivatizing reagents could be adapted to the determination of ammonium and total primary amine groups. In previous studies, we have proposed the use of OPA and the thiol N-acetyl-cisteine (NAC) reagents for the unbiased fluorescent ammonium determination in water samples [9] exciting at 415 nm. Some advantages over ammonium reference methods, such as Nessler reagent method or the ammonium selective electrode method, are the lower detection limit, the absence of any systematic error and amines interference and less toxicity of reagents. Other methods exist for measuring ammonium with (OPA)-derivatization reactions, such as OPA-sulphite reagent [10] or OPA- tioglycolate [11] by forming fluorescent derivatives. Recently we have reported the determination of primary amine groups in water samples, by performing amine derivatization on C18 cartridges and using OPA-NAC reagents [12] and 333 nm as excitation wavelength. The response of several primary amino groups in the OPA-NAC reaction was demonstrated similar in [12], so it was possible to estimate the total primary amine concentration in water employing the calibration graph of methylamine as model compound. By using this solid phase assisted derivatization procedure, ammonia concentrations up to 1.5 mg/L was not an interference species.
- 249. 225 In this paper, however, we have studied the simultaneous determination of ammonium and methylamine as model compound of primary amine groups by using solution derivatization with OPA-NAC reagent, two different excitation wavelengths and a multivariate calibration methodology. When samples are excited at 333 nm, the determination of primary amines or ammonium can be performed measuring the signal at 440 nm. On the other hand, when samples are excited at 415 nm, selective determination of ammonium is performed measuring at 485 nm. Also Principal Component Regression was tested using the spectra of emission obtained exiting at 333 and 415 nm. The method has been applied before and after Kjeldahl sample treatment, and it can be a good indicator of the organic nitrogen present in the sample and if the Kjeldahl procedure fails in the conversion to ammonium of the primary amine groups. The method was applied to several real samples, and the results were compared with the Nessler reference method, obtaining good values. 2.EXPERIMENTAL Apparatus All spectrophotometric and spectrofluorimetric measurements were made on a Hewlet-Packard (Avondale, PA, USA) HP 8452 diode array spectrophotometer furnished with a 1 cm path length, and Hitachi F-4500 fluorescence spectrophotometer, respectively. The pH was measured with a Crison micropH 2000 pH-meter. Digestion Unit for Kjeldahl treatment was a Tecator Digestion system 6, 1007 Digestor (Höganäs, Sweden), and Distillation Unit was a Büchi 323 Distillation Unit (Switzerland). Reagents and standard solutions All solutions were prepared in nanopure water and all reagents were of analytical grade. Stock standard solutions of ammonium were prepared by dissolving ammonium chloride (Probus, Spain) in nanopure water (100 or 1000 mg/L). Standards solutions with variable amounts of ammonium were obtained by dilution. Methylamine (MA) solutions were prepared in the same way. OPA-NAC method: OPA (Fluka chemika, Switzerland)/NAC (Fluka chemika, Switzerland) were prepared with a 1:1 ratio at 8.8mM, dissolving OPA reagent with 5% of MeOH (Scharlau, Spain). Borate buffer (0.5 M), pH 10.8 was prepared by dissolving an adequate amount of boric acid (Scharlau, Sapin) in water and then adjusting the pH with NaOH (Panreac, Spain). Ethylamine (EA), N-Propylamine (N-PrA), Isopropylamine (IPA), Butylamine (BA), Pentylamine (PeA), Hexylamine (HA), Dimethylamine (DMA), Diethylamine (DEA), and β-Phenylethylamine (β-FEA) (Sigma, Germany) solutions at 0.75 mg/L level were prepared. Other concentrations assayed appear in the text. Mixtures
- 250. 226 of amines: 0.75 mg/L EA and 0.75 DEA; 0.5 DMA, 0.5 N-PrA and 0.5 IPA; 0.75 BA and 0.75 PeA; 0.75 β-FEA and 0.75 HA were also used. Nessler method [13]: Sodium-potassium tartrate solution (Panreac, Spain) was prepared by dissolving 50g of the compound in 100 ml of nanopure water and Sodium hydroxide (Panreac, Spain) by dissolving 12g of the compound in 50 ml of nanopure water. Nessler reagent (NR) was prepared by dissolving 3g of mercury chloride (Merck, Germany) in the minimum amount of nanopure water; 10 g of potassium iodine (Guinama, Spain) was diluted up to 50 ml, and then it was added to mercury chloride solution until a red precipitate appears; then the solution was decanted; 18g of sodium hydroxide was diluted up to 100 ml and 40 ml of this solution were added to the HgI4 2- solution; the final solution was diluted up to 100ml. Kjeldahl treatment: For sample or standard digestion, red mercury oxide (Panreac, Barcelona, Spain), potassium sulphate (Prolabo, Fontenay, France) and sulphuric acid (Fluka Chemie, Steinheim, Switzerland) were used. Distillation reagent was prepared by dissolving 25 g of sodium thiosulphate (Prolabo, Fontenay, France) and 500 g of sodium hydroxide (J.T.Baker, Deventer, Holland) together in 1L of nanopure water. Procedures Standard solutions OPA/NAC reaction In a quartz cuvette were placed variable volumes of ammonium and/or amine (alone or mixed) stock solution and nanopure water if necessary up to a volume of 1 mL , 0.1 mL of borate buffer 0.5 M (pH= 10.8) and 0.9 mL of OPA/NAC reagent. The reaction was assumed to start after the addition of the last drop of OPA/NAC reagent. Each experiment was assayed by recording spectra emission between 430-600 nm (λ exc= 415) and 380-610 nm (λ exc = 333) - over the reaction time ranged 0-300 seconds. Signal was obtained at 120 seconds for λex = 333 nm λ em = 440 nm, and at 300 s for λex = 415 nm λ em = 485 nm. All measurements were performed at 25ºC. The ammonium and methylamine concentrations were ranged from 0 to 1.75 mg/L and 0-0.625 mg/L respectively, following a 52 design (Table 1). TABLE 1. Code and concentration (mg/L) of standard calibration set (5 2 design). NH4 + MA (mg/L) 0 0.25 0.375 0.5 0.625 0 00 01 02 03 04 0.25 10 11 12 13 14 0.75 20 21 22 23 24 1.25 30 31 32 33 34 1.75 40 41 42 43 44
- 251. 227 Nessler reaction In a plastic cuvette were placed volumes varied of ammonium stock solution, 10 µL of sodium-potassium tartrate (0.177 M) and water up to constant volume (2.4 mL), 0.1 mL of NaOH 6M and 0.1 mL of Nessler reagent were added to the mixture. The reaction was assumed to start after the addition of the last drop of reagent. Each experiment was assayed by recording spectra between 300-700 nm at 30 s intervals over the reaction time ranged 0-600 seconds. Signal was obtained at 425 nm and 400 seconds. Absorbance signal was measured against water blank. All measurements were performed at 25ºC. Kjeldahl treatment For the application of Kjeldahl treatment [14], several digestion-distillation conditions were assayed (see Table 2). In the digestion step, potassium sulphate amount was varied in the range 2.22-10.5 g, mercury oxide between 0.0332-0.525 g and concentrated sulphuric acid between 3.39-12 mL. A standard volume of 100 mL was added to the mixture and then, the tubes were placed on the Kjeldahl digestor which was previously heated at 370ºC. Heating must be running until white fumes appeared (digestion time near 90 minutes) or must be continue for 30 minutes more (digestion time of 120 minutes). After that, digestion residue must be cooled and diluted up to 50 mL with nanopure water. Distillation must be performed by adding variable volumes (25-50 mL) of distillation reagent (NaOH/Na2S2O3). Distilled ammonia must be picked up on 25 ml of 0.04 N sulphuric acid and the distillation finished when the volume was 80 mL. Collected solutions were diluted up to 100 mL. Before standard procedure the pH of the solutions was adjusted to 10.5. TABLE 2. Different Kjeldahl conditions assayed. Condition m(K2SO4) (g) m(HgO) (g) V(H2SO4) (mL) Digestion Time (min) V(NaOH/Na2S2O3) (mL) 1 2.224 0.0332 3.39 90 25 2 2.224 0.0332 3.39 120 25 3 10.5 0.525 12 120 50 4 6.7 0.1 10.2 120 30 5 6.7 0.1 10 120 40 6 6.7 0.525 10 120 30 Water samples Environmental water samples with unknown ammonium concentration were analysed. They were named as S1: Irrigation ditch water, S2: Residual water from a factory and S3: fountain water.
- 252. 228 Sample treatment The samples or standards were treated in order to eliminate residual chloride and cation interference, according to the reference standard method [15]. A 100 mL portion of water (real or standard) was subjected to sample treatment by adding 1 mL of dechlorant sodium sulphite 7.14 mM and 1 mL of zinc sulphate heptahydrated 0.348M. The formed precipitate was filtered and the first 25 mL of the sample were wasted, collecting the rest of the sample. The pH was adjusted to 2 with concentrated H2SO4 for sample conservation. Sample pH was adjusted to 10.5 before OPA/NAC or Nessler reactions were carried out. OPA/NAC reaction: 1 mL of treated standards or sample for S1 and S3, and 0.1 mL diluted up to 1 mL for S2, were analysed by adding reagents as explained above for standards. Nessler reaction: 2.4 mL of treated standards or sample for S1 and S3, and 0.1 mL diluted up to 2.4 mL for S2, were analysed by adding reagents as explained above for standards. Kjeldahl treatment 100 mL of sample were treated by Kjeldahl method (condition 6, Table 2) for S1 and S3; for S2, 7 mL of sample was diluted up to 100 mL before applying Kjeldahl method. Kjeldahl method was applied in triplicate for each sample. When OPA/NAC reaction was applied, 1 mL of digested sample for S1 and S3, and 0.3 mL of digested sample for S2 diluted up to 1 mL, were used. When Nessler method was applied, the volumes of digested sample employed were 2.4 mL for S1 and S3, and 0.5mL diluted up to 2.4 mL for S2. Calculations The data were aligned according to the maximum emission signal for Principal Component Regression (PCR). For all calculations, Unscrambler (CAMO, Norway) and Matlab for Windows (Math Works, Natick, MA) were used. RESULTS AND DISCUSSION Calibration step According to a previous work [12], standards of Methylamine, Ethylamine, N- Propylamine, Isopropylamine, Butylamine, Pentylamine, Hexylamine, and β- phenylethylamine provided similar response between them when concentration was expressed in mg/L of Nitrogen, with %Recovery respect to methylamine signal of 100, 93.5, 101.1, 93.4, 89, 100.4, 108.2 and 99 % for each amine, respectively. The same emission maxima located at 440 nm were also obtained for all of them exciting at 333 nm (Figure 1). The additivity between different amine signals was tested analysing mixtures of amines (see experimental section). %Recoveries obtained were between 81-88%. With these results, it was corroborated that the amine response in the OPA-NAC reaction was not dependent on the primary amine, and methylamine could be used as a representative compound.
- 253. 229 FIGURE 1. Fluorescence emission spectra for a standard of 1.75 mg/L of ammonium and a standard of 0.375 mg/L of methylamine, both at λexc =333 nm and at λexc=415 nm. In order to study the simultaneous determination of ammonium and primary amines (c.a. methylamine), a design 52 was assayed (Table 1). Both, Ammonium and primary amine, provide fluorescent signal measuring at λem 440 nm if λex was 333 nm, although the emission maximum for ammonium was 462 nm (Figure 1). There were not significative differences at 95 % confidence level (homogenous variance, Fcal<Ftab-α=0.05, and comparable slopes, tcal<ttab-α=0.05) between the amine calibration graphs obtained with different constant amounts of ammonium ranged from 0 to 1.75 mg/L (Table 3). The contribution of the ammonium to the total signal can be seen in the corresponding ordinate values. This fact corroborates the additively of ammonium and amine signals. The same is true for ammonium slopes and ordinates obtained in presence of different amine amounts (Table 3). The mean slopes obtained at 440 nm for amine and ammonium were 6532 (s= 472, n= 5) and 1038 (s= 127, n= 5) and expressed considering N 2950 (s= 213, n= 5) and 855 (s= 104, n= 5), respectively. These values indicated that the amine isoindols are 3.5 times more sensitive than ammonium isoindol exciting at 333 nm and measuring at 440 nm. Figure 1 shows the registers obtained for ammonium and methylamine exciting at 415 nm. Blank signal was obtained for the amine and a maximum at 485 nm for ammonium. As can be seen in Table 3 at the selective excitation wavelength for ammonium, that is λex 415 nm (λem 485 nm) the same calibration graph was obtained in presence or absence of amine. For different amounts of amine present the ordinates were similar to that provided by the ammonium calibration graph, its contribution being due to the blank reagent. The ammonium calibration graph at 485 nm and the ammonium and amine calibration equations at 440 nm, exciting at 415 and 333 nm respectively, were needed to estimate both analytes as demonstrated in the following. 0 500 1000 1500 2000 2500 3000 3500 4000 380 430 480 530 580 λλ (nm) FluorescenceSignal MA λexc = 333 nm NH4 + λexc = 333 nm NH4 + λexc = 415 nm MA λexc = 415 nm
- 255. 231 The obtained emission spectra exciting at 415 and 333 nm for the standards (Table 1) were processed by using principal component regression (PCR) in order to develop a multivariate model to ammonium and amine, respectively. Top-Down selection (TDS) and Best-Subset selection (BSS) have been employed to perform the calibration step [16]. PCR models were obtained using as X-block the data (column centred). The number of variables, the percentage of explained variance, the number of factors and root mean squared errors of prediction (RMSECV) by leave-one-out cross- validation are given in Table 4. RMSECV values were calculated in order to evaluate the prediction ability of models. For methylamine there are no differences between the two options of selection, and a model with complexity three has been selected. For ammonium PC number 2 is not included in the model with BSS selection, and only PCs number 1 and 3 have been used (complexity two). In a second step, variable selection on the optimised model by using the Uninformative Variable Elimination (UVE) approach has been used [17]. Only 32 and 29 original variables have been used for methylamine and ammonium, respectively. In both cases a final model complexity two has been selected. The RMSECV values obtained are good as can be seen in Table 4 for all models, although UVE-BSS models were selected. TABLE 4. Characteristics of Principal Component Regression Models obtained for Ammonium and Methylamine by using Top-Down Selection, Best Subset Selection or Uninformative Variable Elimination-Best Subset Selection. Ammonium (ëexcitation 415 nm) Amine (ëexcitation 333 nm) TD BSS UVE-BSS TD BSS UVE-BSS Number of variables 231 231 32 180 180 29 Selected PCs 1,2 1,3 1,2 1,2,3 1,2,3 1,2 Explained variance, block X 99.999 99.999 99.999 99.999 99.999 99.999 rmsecv 0.089 0.051 0.052 0.034 0.037 0.040 Accuracy and precision Taking into account previous results, the standards were analysed. The measurements were performed exciting at 415 and 333 nm and using the ammonium calibration graph at 485 nm and the ammonium and amine calibration equations at 440 nm, respectively. Table 5 shows the ammonium and amine concentration found in the different mixtures assayed. Satisfactory results were obtained in all cases with relative errors for amine and ammonium contents between 7-8 % and 4-24 % and relative standard deviations between 6-13 % and 1-9 %, respectively. Te same mixtures were analysed by using the PCR model. Improved results were obtained for accuracy of ammonium. The relative errors found were between 1-16 %.
- 256. 232 TABLE 5. Found amine and ammonium concentration in standard solutions (mixtures of ammonium and Methylamine) and real samples by applying bivariate calibration graphs with standards or PCR. Found ammonium (mg/L) Found amine (methylamine) (mg/L)Samples Added NH4 + (mg/L) Bivariate Calibration PCR Added MA (mg/L) Bivariate Calibration PCR 0.25 0.31 ± 0.03 0.29 ± 0.02 0.25 0.23 ± 0.03 0.26 ± 0.02 0.75 0.89 ± 0.04 0.76 ± 0.06 0.375 0.35 ± 0.02 0.40 ± 0.06 1.25 1.14 ± 0.03 1.23 ± 0.07 0.5 0.46 ± 0.06 0.50 ± 0.04 Standard 1.75 1.68 ± 0.02 1.74 ± 0.06 0.625 0.58 ± 0.04 0.58 ± 0.09 Sample 1 n.d n.d. 0.051 ± 0.007 0.086 ± 0.009 Sample 2 24.92 ± 0.10 32.2 ± 0.4 8.8 ± 0.4 10.25 ± 0.14 Sample 3 0.36 ± 0.03 0.29 ± 0.05 0.058 ± 0.013 0.082 ± 0.004 Kjeldahl method Different Kjeldahl conditions were assayed in order to select the best, which guarantee the total amine conversion (Table 2). The variables assayed were: HgO amount (g) (0.0332 to 0.525 g), concentrated H2SO4 volume (3.39-12 mL), K2SO4 amount (2.22- 10.5g), digestion time (90-120 minutes), distillation reagent volume NaOH/Na2S2O3 (25- 50 ml). The OPA/NAC proposed method was used for testing conversion by use of the bivariate calibration model. While the ammonium extraction from the sample was almost independent of the experimental conditions (Figure 2), the amine transformation into ammonium was dependent as can be seen in Figure 3. Condition 6 provided a total transformation of MA into ammonium. The methylamine conversion into ammonium was 109 % . A similar value was obtained by the Nessler method applying condition 6. 0 20 40 60 80 100 120 140 160 2 3 5 6 Condition %Recovery 440 nm 485 nm FIGURE 2. %Rec of NH4 + calculated regarding to signal (or slope) obtained without Kjeldahl treatment, at different Kjeldahl conditions (Table 2). Cond 2: n=7 : 5 st of 0.5 mg/L of NH4 + and 2 calib.graphs (0, 0.5, 1.25, 2, 2.75 mg/L NH4 + and 0, 0.2, 0.5, 0.9, 1.3 mg/L NH4 + respectively). Cond. 3: n=4: 2 st. of 0.5 mg/L of NH4 + and 2 calib.graphs (0, 0.05, 0.25, 0.78 mg/L NH4 + , and 0, 0.75, 0.75 mg/L NH4 + ). Cond. 5: n=1: Calib.graph (0, 0.28, 0.56 mg/L NH4 + ). Cond. 6: n=3: St. of 0.375 and 0.75 (2 replicates) mg/L NH4 + .
- 257. 233 FIGURE 3. %Recovery of MA transformed into ammonium and %Recovery of MA not transformed, calculated regarding to signal obtained without Kjeldahl treatment, at different Kjeldahl conditions (Table 2). Condition 1: n=4 : 2 standards of MA of 0.5 mg/L and 2 standards of 0.125 mg/L. Condition 2: n=4: Standards of MA of 0.086, 0.3875, 0.5 and 0.56 mg/L. Condition 4: n=2: Standard of MA of 0.5 mg/L (2 replicates). Condition 5: n=5: Standard of 0.775 mg/L of MA (5 replicates). Condition 6: n=2: Standard of 1.3 mg/L of MA (2 replicates). Other primary and secondary amines, such as ethylamine, n-propylamine, isopropylamine, butylamine, pentylamine, hexylamine, dimetylamine, diethylamine and β-phenylethylamine, were also assayed in order to study its conversion into ammonium. In Figure 4 are shown the fluorescence spectrum at λexc 415 of the OPA-NAC- ammonium and amine derivatives at different concentration levels before Kjeldahl and ammonium concentration of 0.75 mg/L after Kjeldahl treatment and OPA-NAC derivatization. As can be seen the analytical signal of the amine derivatives before Kjeldahl digestion is similar to the blank (Figure 4A). However, after performing the Kjeldahl digestion the amines are converted into ammonium (Figure 4B). The ammonium measurement was performed by OPA-NAC procedure (λexc 415 - λem 485 nm) and contrasted by the Nessler method; the results are shown in Table 6. As can be seen, comparable results were provided by both methods, and recoveries nearly 100% were obtained in solutions with only one amine, as well as in amine mixture standard solutions. 0 20 40 60 80 100 120 140 1 2 4 5 6 Condition %Recovery MA 440 nm NH 485 nm
- 258. 234 FIGURE 4. Fluorescence emission spectra corresponding to different OPA-NAC isoindol derivates at 485 nm (λexc= 415 nm) for blank, standard of 0.75 mg/L NH4 + , 1.3 mg/L MA, 1.875 mg/L EA and DMA, 2.465 mg/L N-PrA and IPA, 3.05 mg/L BA and DEA, 3.625 mg/L PeA, 4.215 mg/L HA, and 5.05 mg/L β-FEA. (4A)-measured before Kjeldahl digestion. (4B) measured after Kjeldahl digestion. 450 500 550 600 0 200 400 600 800 1000 FluorescenceSignal λem (nm) Standard 0.75 mg/l NH4+ Blank, M A, EA, N PrA, IPA, BA, PeA, HA, DMA, DEA, B-FEA A) 450 500 550 600 0 200 400 600 800 1000 D MA BA H A, NPrA St 0.75 m g/l N H4+, M A, EA, IPA, PeA, DEA, B-FEA B) FluorescenceSignal λem (nm) Blank
- 259. 235 TABLE 6. % Ammonium recovery (n=1) of different amine after Kjeldahl digestion, by applying Nessler or OPA-NAC method at 485 nm. (EA-ethylamine, N-PrA–N- propylamine, IPA-iso-propylamine, BA-butylamine, PeA-Pentylamine, HA-Hexylamine, DMA-Dimethylamine, β-PEA-β-Phenylethylamine). Amine (Concentration before Kjeldahl digestion) OPA-NAC Method (%ammonium recovery) Nessler Method (%ammonium recovery) EA (3.75 mg/L) 101 110 N-PrA (4.93 mg/L) 122.5 121.5 IPA (4.93 mg/L) 107.2 103.13 BA (6.10 mg/L) 88.2 81.8 PeA (7.25 mg/L) 99.8 96 HA (8.43 mg/L) 124.7 121 DMA(3.76mg/L) 71.5 68.6 DEA(6.10 mg/L) 112 101.2 β-FEA(10.10 mg/L) 104.4 100.7 EA (1.878 mg/L) + DEA (3.047 mg/L) 84.4 79 DMA (1.253 mg/L) + N- PrA (1.642 mg/L) + IPA (1.642 mg/L) 82 81 BA (3.05 mg/L) + β−FEA (5.05 mg/L) 99.9 85 PeA (3.622 mg/L) + HA (4.21 mg/L) 85.3 92 Application to real samples The procedure has been applied to the determination of ammonium and primary amines (expressed as methylamine) in real water samples (see experimental section). Three water samples of different nature were analysed: Ditch water, Industrial waste water and Fountain water. Based on the results showed in Table 5, a t test of related samples gave á > 0.05 for both ammonium and methylamine determination. Therefore, similar results were obtained by using both calibration systems, bivariate and PCR models. Sample 3 was also analysed by Nessler method and comparable results for ammonium were obtained: 0.32 ± 0.02 (n= 3). The OPA-NAC procedure was applied to the water samples after Kjeldahl digestion. In this case, the analytical measurement of ammonium corresponds to the ammonium content plus the organic nitrogen. The results obtained have been compared with those obtained by Nessler method. The results of ammonium found were (0.19 ±
- 260. 236 0.07), (213 ± 3) and (2.103 ± 0.018) mg/L ammonium for ditch, industrial waste and fountain waters, respectively, which were similar to those obtained by Nessler (t test for paired samples á > 0.05): (0.223 ± 0.018), (232 ± 3), (2.14 ± 0.02) mg/l ammonium respectively. These results are in agreement of the water nature, for natural water with low pollution, the N-Kjeldahl will not exceed 5 mg/l. For industrial waste water, this parameter can be higher than 200 mg/L of ammonium. Bearing in mind the results of Table 5, other amino compounds besides ammonium and primary aliphatic amines, are present in these waters. Conclusions In this article, the use of OPA/NAC reagent for the simultaneous determination of ammonium and primary aliphatic amines has been proposed. Bivariate calibration or multivariate Principal Component Regression can be used obtaining similar results. Methylamine can be used as model compound, because the response of each amine was the same when it was expressed in mg/L of Nitrogen, and there was additivity in their signal when mixtures were processed. Similar calibration curves were obtained for ammonium (0.25-1.75 mg/L) with a fixed amine concentration ranged from 0.25 to 0.625 mg/l methylamine exciting at 415 nm. No response for amines was obtained working at this wavelength. Slopes of the Methylamine calibration curves (0.25-0.625 mg/l) were also similar with a fixed ammonium concentration ranged from 0.25 to 1.75 mg/l exiting at 333 nm. The signal of the ammonium affecting ordinate values. Kjeldahl treatment was applied obtaining similar slopes for ammonium calibration graphs before and after the treatment. Amine conversion into ammonium was near 100% for every amine studied alone or in mixtures. The application of Kjeldahl treatment to real water samples gave results in agreement of the water nature, and Nessler method was applied in order to validate accuracy of the proposed method, providing similar results to those obtained by the OPA/NAC method. Acknowledgements The authors are grateful to the Ministerio de Ciencia y Tecnología of Spain for financial support received for the realization of Project PPQ 2000-1641. S.M.Ll. expresses her gratitude to Ministerio de Educación y Cultura (Spain) for the predoctoral grant. References [1] A. Aminot, D.S. Kirkwood and R. Kérouel. Mar.Chem. 56 (1997) 59. [2] C.Dodeigne, L.Thunus, R.Lejeune. Talanta, 51 (2000) 415. [3] D.Sahasrabuddhey, A.Jain, K.K.Verma. Analyst, 124 (1999) 1017.
- 261. 237 [4] M. Kaminski, D.Jastrzebski, A.Przyjazny, R.Kartanowicz. J.Chromatogr. A, 947 (2002) 217. [5] F. Dai, V.P. Burkert, H. N. Singh and W.L. Hinze. Microchem. J. 57 (1997) 166. [6] P. Campíns-Falcó, C. Molins-Legua, A. Sevillano-Cabeza and L. A. Tortajada Genaro, J. Chromatogr. B. 759 (2001) 285. [7] H.Mana, U.Spohn. Fresenius J.Anal.Chem. 366 (2000) 825-829. [8] A.Aminot, R.Kerouel, D.Birot. Wat.Res. 35 (7) (2001) 1777-1785. [9] S.Meseguer-Lloret, C.Molins-Legua, P.Campins-Falcó. Intern.J.Envir.Anal.Chem. 82 (7) (2002) 475-489. [10] A. Aminot, R.Kerouel, D.Birot, Wat.Res. 35-7 (2001) 1777-1785 [11] H.Mana and V.Spohn, Fresenius J Anal Chem, 366, 825-829 (2000). [12] A.Sevillano Cabeza, Y.Moliner Martinez, C.Molins Legua, P.Campíns Falcó. Anal.Bioanal.Chem. 376 (2003) 918-922. [13] F. Burriel. Química analítica Cualitativa, 17th Edn., Pub Paraninfo, Madrid (2000). [14] Application Note ASN 52/85. Tecator Manual Digestion system 1007 (1985). [15] S.A.Díaz-Santos. Métodos Normalizados para el análisis de aguas potables y residuales, 17th Edn., Apha-Awwe-WPCF, Madrid (1992). [16] J.Verdú, D.L.Massart. Applic. Spectrosc. 52 (1998) 1425-1434. [17] V.Centner, D.L.Massart, O.E. de Noord, S.de Jong, B.M.Vandeginste and C.Stena. Anal.Chem. 68 (1996) 3851.
- 262. 238
- 263. 239 Apéndice 11. UNPUBLISHED PAPER CHEMILUMINESCENCE AUTOMATIC METHOD FOR TOTAL ESTIMATION OF ORGANIC NITROGEN (-CH2-NH-) AND AMMONIUM S. Meseguer Lloret, C. Molins Legua, J. Verdú Andrés and P.Campíns Falcó* Departament de Química Analítica, Facultad de Química, Universitat de Valencia C/ Dr. Moliner 50, E46100- Burjassot, Valencia. SPAIN Abstract An automatic method has been developed for estimation of organic nitrogen (- CH2-NH-) and ammonium in water samples. We proposed a continuous flow system in which nitrogen compound reacts with hypochlorite reagent to form chloramines and, subsequently, the mixture was merged with luminol, generating a chemiluminescence signal. The signal emission at 425 nm, registered as a function of time, decreases as nitrogen concentration increases, due to the decrease on hypochlorite concentration. A large number of nitrogen compounds has been assayed and sensitivities compared, in mg/L of Nitrogen. For all of them the same calibration graph was obtained. The linear interval was 0.24- 4 mg/L of N, being the detection limit 0.07 mg/L of N. The chemiluminescence method was applied to the analysis of several kind of real water samples: natural water, lake water, irrigation ditch water, fountain water, residual water and seawater. A comparative study of ammonium nitrogen contents before and after Kjeldahl treatment has been carried out. Except for two samples, the N found for the untreated samples provided a good estimation of the N Kjeldahl. 60 samples per hour can be analysed and the procedure used also for in situ monitoring. Keywords: ammonium, organic nitrogen (-CH2-NH-), water samples, chemiluminescence, luminol, hypochlorite, Total Kjeldahl Nitrogen. * Correspondence author. E-mail address: pilar.campins@uv.es (P.Campins).
- 264. 240 INTRODUCTION Nitrogen is a key nutrient in natural waters, but excess N inputs lead to eutrophication. The dominant N species in waters are: dissolved inorganic N (ammonium, nitrite and nitrate), dissolved organic N (the largest fraction is made up of amino acids and peptides and it is often called amino N) and particulate organic (due to small organisms: algae, bacteria) and inorganic N [1]. Inputs of dissolved organic nitrogen (DON) to natural waters are largely a result of autochthonous biological processes. Additionally, external sources of DON arise from sewage and industrial effluents, terrestrial run-off and atmospheric deposition [2]. DON content in water has largely been ignored, difficulties in measuring DON, its chemical character remains poorly described and an underlying assumption that it is biologically inert can be the causes [2]. Some analytical procedures exist for quantifying dissolved inorganic N (DIN), total dissolved nitrogen (TDN) but not for dissolved organic N as mentioned above. The most used is the Kjeldahl wet chemical method [3-6] because it is the reference method established by European Directive [7] relative to measurement methods in the analysis of superficial waters. Kjeldahl method was developed specifically for proteinaceous nitrogen and some authors indicated incomplete recovery of nitrogen for some compounds [8,9]. It provides DON and ammonium contents. In other protocols DON is calculated as the difference between independent measurements of TDN and DIN, but they are even more time-consuming than Kjeldahl method. The main methods for TDN determinations are: alkaline persulfate oxidation or UV-photo-oxidation to nitrate and oxidative combustion methods to NO [2,8]. Some papers have been published comparing several of the used methods [2,10,11]. More work is needed in order to improve accuracy of DON results and reduce time and/or difficulty of the technique. Based on previous studies for ammonium [12] and the known reaction between amine groups and hypochlorite, we propose a continuous flow system for organic amino N and ammonium nitrogen determination on untreated water samples, in which amino group react with hypochlorite reagent to form a chloramine and, subsequently, the mixture reacts with luminol, generating a chemiluminescence signal. The signal emission at 425 nm, registered as a function of time, decreases as nitrogen concentration increases, due to a decreasement on hypochlorite concentration that depends on the amino group present in the sample. A large number of nitrogen compounds has been assayed and sensitivities compared, in mg/L of Nitrogen. A unique calibration graph can be used for a great number of compounds tested and then, the method can be employed for estimating the main dissolved organic nitrogen and ammonium. Characteristics of the method, such as selectivity and reproducibility, have been evaluated. The chemiluminescence method was applied to the analysis of several kind of real water samples (natural water, lake water, irrigation ditch water, fountain water, residual water and seawater). A comparative study of ammonium nitrogen contents before and after Kjeldahl treatment has been carried out. Standard additions method was
- 265. 241 applied for all samples studied in order to test the presence or absence of proportional systematic errors. EXPERIMENTAL Apparatus All spectrofluorimetric measurements were made on a Hitachi F-4500, 700v fluorescence spectrophotometer (Tokio, Japan). The light emission was monitored at 425 nm. The pH was measured with a Crison micropH 2000 pH-meter. A Nanopure II (Synbron Barnstead) water unit was used. For FI assembly (see Figure 1), a Gilson Miliplus peristaltic pump was used to drive the reactants through the flow cell; it always worked at a flow rate of 4.1 ml/min. A 400 µL reactor was employed to perform the reaction between Hypochlorite and Nitrogen compounds. The sample loop employed had a 50 µL internal volume. Tygon tubing (i.d. 0.8 mm) was used with the peristaltic pump. The other tubing was made of PTFE (i.d. 0.5 mm). The flow cell was a laboratory-made bundle cell [13], consisting of knotted transparent poly-tetrafluoroethylene tube, measuring 50 cm in length and 0.8 mm of internal diameter. The bundle cell (1.5x1x1 cm) was placed in a plastic cell. A second kind of flow cell studied was a laboratory-made spiral cell [13] consisting of coiled transparent PTFE tube measuring 50 cm in length and 0.8 mm of internal diameter. Its dimensions were 1 cm (internal diameter) and 3 cm (external diameter). For sample Kjeldahl treatment, the Digestion Unit was a Tecator Digestion system 6, 1007 Digestor (Höganäs, Sweden), and the Distillation Unit was a Büchi 323 Distillation Unit (Switzerland). Reagents and solutions All solutions were prepared in nanopure water, and all reagents were of analytical grade. Stock standard solutions of ammonium were prepared by dissolving ammonium chloride (Probus, Barcelona, Spain) in nanopure water. The following amines from Sigma (Steinheim, Germany) were employed: methylamine (MA), ethylamine (EA), n-propylamine (nPA), isopropylamine (iPA), butylamine (BA), pentylamine (PeA), hexylamine (HA), dimethylamine (DMA), diethylamine (DEA), trimethylamine (TMA), triethylamine (TEA), β-phenylethylamine (BPEA), 1,7-diaminoheptane (DAH), putrescine (Put), cadaverine (Cad), spermine (Spm) and spermidine (Spd). The following aminoacids from Aldrich (Steinheim, Germany) were employed: aspartic acid, tryptophan, glutamic acid, glicocole, proline, serine, tyrosine, alanine, phenylalanine, valine, leucine, isoleucine, treonine, lisine, metionine, histidine, cysteine, histidine and arginine. Other solutions (1000 mg/L) were prepared: Mercaptoethanol (Scharlau, Barcelona, Spain); Urea and sodium sulphide from Prolabo, Fontenay, France; N-Acetyl- L-Cysteine, s-Triazine and albumine from bovine serum were from Fluka (Switzerland);
- 266. 242 Moreover, metal solutions (0.05 M) of chromium chloride, cobalt (II) chloride, copper (II) chloride, iron (III) chloride, cadmium chloride and mercury (II) chloride from Panreac (Barcelona, Spain) were prepared. The following reagents were also used: sodium hypochlorite 15%, sodium thiosulphite and potassium sulphate from Prolabo (Fontenay, France); sodium hydroxide, sulphuric acid and sodium carbonate from Merck (Darmstadt, Germany), and red mercury oxide from Panreac (Barcelona, Spain). PROCEDURES FI procedure The flow injection assembly is shown on Figure 1. Initial concentrations and flow injection system assembly were the ones described in [12], with some modifications. Firstly, hypochlorite was not electrogenerated, in order to have a simpler system. Secondly, the sample was previously mixed with hypochlorite in a reaction coil of 400 µL in order to form the chloramine compound and, then, sample loop was filled up. A water carrier picked up the loop contents when injection valve was turned, and it was mixed with luminol / buffer carrier. The distance between the last T-junction and the detection cell was the minimum possible, 5 cm. The working solutions were as follows: hypochlorite 0.9 mM (in hydrogencarbonate / carbonate buffer 0.144 mM, pH 11.2), and luminol 1 mM (in hydrogencarbonate / carbonate buffer 0.2 M, pH 11.2). FIGURE 1. Flow injection assembly for chemiluminescence organic nitrogen determination. The following standard solutions were prepared and measured according to the methodology proposed: Standard solutions of ammonium, amines, amino acids, N-acetyl-L-cysteine (NAC), s-triazine and urea were measured in the range of 0-4 mg/L of nitrogen. Albumine from bovine serum was measured in the range of 2.5-100 mg/L of albumine. W Detection cell Sample loop: 50 µL IV Reactor, 400 µL Standard/Sample H2O Luminol 1 mM, 0.2M CO3 2- /HCO3 - pH 11.2 Hypochloride 9 µM, 0.144mM CO3 2- /HCO3 - pH 11.2 v = 35 rpm W
- 267. 243 Standards of cysteine, metionine, NAC, mercaptoethanol and sodium sulphide were prepared in the range of 0-5.4· 10-5 M of sulphide. Metal solutions of: Copper (II) (2· 10-6 , 4· 10-5 and 5· 10-4 M), Fe(III) (9· 10-6 , 5· 10-4 and 5· 10-3 M), Cd(II) (4.5· 10-8 , 2.5· 10-4 and 2.5· 10-3 M), Hg(II) (5· 10-9 , 6.25· 10-3 and 0.05M), Cr(III) (1· 10-5 M) and Co(II) (8.5· 10-6 M) were also measured. Kjeldahl treatment For the application of Kjeldahl treatment [14], 6.7 g of potassium sulphate, 0.52 g of red mercury oxide and 10 mL of concentrated sulphuric acid, were added to 100 mL of nitrogen containing compound standard in a digestion tube. Then, the tubes were placed on the Kjeldahl digestor, previously heated at 370ºC. Heating is kept until white fumes appeared and, then, digestion continues for 30 minutes more. After that, digestion residue must be cooled and diluted up to 100 mL with nanopure water. Distillation is performed after the addition of 35 mL of distillation reagent (25 g of sodium thiosulphate and 500 g of sodium hydroxide diluted up to 1L with nanopure water). Distilled ammonia must be picked up on 25 mL of 0.04 N sulphuric acid until 80 mL. This solution was diluted up to 100 mL. Before experimental measures, collected solutions were adjusted to pH 6.0-7.0. Solutions (100 mL) of TEA, urea, histidine and arginine, with final concentrations of 1.5 mg/L of nitrogen, solution mixtures of MA / PeA / DMA or MA / PeA / DMA / TEA, with final Nitrogen concentration of 1.5 mg/L or 1.8 mg/L respectively, and a solution with 1 g/L of albumine, were submitted to Kjeldahl treatment. Sample procedures Eight different water samples were collected and pH adjusted to 2.0 with concentrated nitric acid until they were used. At the moment of the analysis, the samples pH value was adjusted near 7.0. All of them were analysed directly by the proposed FI chemiluminescence method, before and after the Kjeldahl treatment. Samples were named as follows: S1: mineral bottled water; S2: tap water; S3 and S7: irrigation ditch water (from different sources); S4: lake water; S5: sea water; S6: industrial waste water and S8: fountain water with clear aspect of eutrophication. 4.45 mL diluted up to 5 mL for S1, S2, S3, S4, S7 and S8, 1ml diluted up to 5 mL for S5, or 0.035 mL diluted up to 5 mL for S6, were prepared and measured in triplicate. These samples were also spiked with ammonium (0.75, 1.5 and 3 mg/L of nitrogen), in order to apply the standard additions method. Kjeldahl treatment Kjeldahl treatment was applied, as described above for standards, to 100 mL of S3, S4, S5 and S8 (all in triplicate), and to 7 mL of S6 diluted up to 100 mL (in triplicate). After Kjeldahl treatment, 4.45 mL diluted up to 5 mL for S3, S4, S5 and S8, or 0.35 mL diluted up to 5 mL for S6, were measured in triplicate. They were also spiked with ammonium (0.75, 1.5 and 3 mg/L of nitrogen), to apply the standard additions method.
- 268. 244 RESULTS AND DISCUSSION Analytical parameters The parameters for the calibration curves chemiluminescence signal vs nitrogen concentration (mg/L), and other figures of merit, for the ammonium determination at different experimental conditions assayed can be seen in Table 1. The different variables tested were: kind of measurement cell, buffer concentration, hyplochlorite concentration, presence of buffer in standards and presence of NaCl in standards. TABLE 1. Ammonium calibration curves (chemiluminescence signal vs nitrogen concentration, mg/L) and other figures of merit for the different experimental conditions assayed. (a: ordinate, sa: estándar deviation of ordinate, b: slope, sb: estándar deviation of slope, n: number of standards, r 2 : regression coefficient, sy/x: standard deviation of calibration curve). Conditions assayed Detection Cell Hypochl. Conc. (mM) Buffer in Hypochl. Buffer in standard solutions NaCl in standard solutions Calibration curve Y = (a ±± sa) + (b ±± sb)· C (n, r2 ,sy/x) DL (mg/l) Linear Interval (mg/l) Spiral 0.9 0.144 mM No No Y = (9170 ± 30) + (-1844 ± 14)· C (12, 0.9994, 70) 0.091 0.3-4 Bundle 0.9 0.144 mM No No Y = (9680 ±± 140) +(-2370 ±± 70)· C (10, 0.9938, 300) 0.070 0.24-4 Bundle 0.5 0.144 mM No No Y = (4300 ± 50) + (-1960 ± 40)· C (9, 0.9968, 100) 0.086 0.3-2 Bundle 0.9 0 mM No No Y = (9690 ± 100) +(-2290 ± 40)· C (12, 0.9962, 200) 0.073 0.24-4 Bundle 0.9 10 mM No No Y = (9400 ± 90) + (-1730 ± 40)· C (12, 0.9940, 200) 0.097 0.3-4 Bundle 0.9 0.144 mM Yes No Y = (7960 ± 160) + (-1570 ±70)· C (12, 0.9806, 400) 0.107 0.4-4 Bundle 0.9 0.144 mM No Yes Y = (8290 ± 100) + (-2050 ±40)· C (15, 0.9942, 200) 0.082 0.27-4 Because the presence of the analyte decresases the chemiluminescence signal, it is interesting to reach almost 10000 arbitrary units for the blank (saturation signal), in order to have the maximum working range for the analyte. The optimum conditions have been marked in bold in Table 1: bundle cell and hypochlorite 0.9 mM in carbonate buffer (0.144 mM, pH 11.2). Good linearity was obtained, and the detection limit reached was 0.07 ppm (referred to nitrogen content in the sample). Sensitivity decreased about 33% when standards were conditioned with buffer, and about 13% when they were prepared in NaCl. FI registers obtained for ammonium calibration standards are shown in Figure 2.
- 269. 245 FIGURE 2. FI registers obtained under optimum experimental conditions at different Nitrogen concentration levels. Conditions: Hypchlorite 0.9 mM in carbonate buffer 0.144 mM pH 11.2 with bundle cell. The reproducibility of the method was measured in two ways. Figure 3 shows how ammonium calibration curves obtained for five different days overlap at every concentration level. The percentage of residual standard deviation (%RSD) for calibration slope was 1.86%. FIGURE 3. Nitrogen calibration curves (Chemiluminescence versus Nitrogen concentration (mg/L)) obtained in five different days. Conditions: Hypchlorite 0.9 mM in carbonate buffer 0.144 mM pH 11.2 with bundle cell. y = -2293.3x + 9365.4 y = -2255.9x + 9570.4 y = -2332.4x + 9456 y = -2339.4x + 9677.6 y = -2366.3x + 9676 0 2000 4000 6000 8000 10000 12000 0 1 2 3 4 5 Nitrogen Concentration (mg/L) Chemiluminescence 26/06/03 30/06/03 01/07/03 02/07/03 25/06/03 Nitrogen calibration curves 0 100 200 300 400 0 2000 4000 6000 8000 10000 ChemiluminescenceSignal Time (s) 0.75 mg/l N BlanK 2 mg/l N 3 mg/l N 4 mg/l N
- 270. 246 On the other hand, repeatibility studies gave a RSD (n=3) of 0.81%, 0.31% and 3.7% for nitrogen concentrations of 0.75, 2 and 4 mg/L respectively. Both values indicate that the precision of the method can be considered rather satisfactory. 60 samples per hour could be analysed without previous treatment and the procedure can be adapted as an in situ monitoring analyser. Response to other nitrogen-containing compounds In order to check the behaviour of the proposed methodology for the amino N determination (organic nitrogen plus ammonium nitrogen), the results for other families of nitrogen-containing compounds were obtained, and compared with those obtained for ammonium (all referred to nitrogen content). The first family of compounds studied were the aliphatic amines: primary, secondary and tertiary. Seven aliphatic primary amines (methylamine, ethylamine, n-propylamine, isopropylamine, butylamine, pentylamine and hexylamine) were processed, and the corresponding recovery results (calculated as slope of amine calibration * 100 / slope of ammonium calibration) are shown in Table 2. The mean recovery obtained, (91 ± 11) %, showed how the primary amine responses were similar to the ammonium response and, for this reason, their nitrogen content could be also measured with this methodology. TABLE 2. Percentage of recovery (slopeamine*100/slopeNH4+) found for primary and secondary aliphatic amines individually and mean %recovery. AMINE % Recovery = bamine*100/bNH4+ Mean Recovery (% REC ± s) methylamine 104 ethylamine 83 n-propylamine 79 isopropylamine 82 butylamine 82 pentylamine 102 hexylamine 103 (91 ± 11)% dimethylamine 78 diethylamine 84 (81 ± 4)% Two aliphatic secondary amines (dimethylamine and diethylmine) were also processed, and the results are also shown in Table 2. Although the mean recovery was slightly lower than for primary amines, (81 ± 4)%, their nitrogen content can be also measured with this methodology. The results for two aliphatic tertiary amines (trimethylamine and triethylamine) showed that this kind of compounds can not be detected with this methodology. This can
- 271. 247 be explained because in these experimental conditions the corresponding chloramine is not formed. Thus, the hypochlorite reagent is not consumed, and the chemiluminiscence signal remains constant. A second family of compounds studied were the polyamines. These molecules contain several amino groups, and thus, the response for each analyte should decrease as many times as amino groups are present. When the calibration is performed against the nitrogen content for five of these compounds (putrescine, cadaverine, spermine, spermidine and 1,7-diaminoheptane), excellent percentage recoveries were found (Table 3). The mean recovery, (96.6 ± 1.8) %, allows the nitrogen content for these kind of compounds to be obtained by applying this methodology. TABLE 3. Percentage of recovery (slopepolyamine*100/slopeNH4+) found for polyamines individually and mean %recovery. POLYAMINE % Recovery bamine*100/bNH4+ Mean Recovery (% REC ± s) putrescine 98 cadaverine 95 spermine 98 spermidine 94 1,7-diaminoheptane 97 (96.6 ± 1.8)% The third family of nitrogen containing compounds studied were arylalkyl amines. â-phenylethylamine was selected as a model of compound. The obtained recovery was 100.1 %, indicating that also the nitrogen content for this kind of amines can be measured with the proposed methodology. Another two types of nitrogen compounds were studied: those containing an aromatic N atom (for example, triazine), and those containing resonant nitrogen (for example, urea). In both cases the chemiluminiscence signal remains constant for all concentrations tested. These results show that aromatic and resonant nitrogen will not be detected by this methodology, however the aromatic nitrogen exalted the blank chemiluminescence signal. In addition, 14 amino acids (with only one amino group) were also studied. The recoveries obtained (Table 4), were near 100% in all cases. The mean recovery was (96 ± 4) % for their nitrogen content, and it was in excellent agreement with the real one. Histidine presents a structure with two aromatic nitrogens besides the amino group and accordly with results for triazine, the chemiluminescence of the blank was increased and no decrease of the signal was observed in the N working range. Arginine presents resonant nitrogens besides the amino group, the obtained results considering only the aliphatic nitrogen (baminoacid* 100/ bammonium= 96.8) confirm that only the –CH2-NH2 group for arginine lowers the chemiluminiscence signal. Cisteine and metionine gave (baminoacid* 100/ bammonium) values of 287 and 147 %. Those recoveries indicate that other atom of
- 272. 248 their structure were contributing to the decrease in chemiluminescence. As can be demonstrated below this atom was S. TABLE 4. Percentage of recovery (slopeaminoacid*100/slopeNH4+) found for aminoacids individually and mean %recovery. AMINOACID % Recovery bamine*100/bNH4+ Mean Recovery (% REC ± s) aspartic acid 93.5 tryptophan 87.1 glutamic acid 101.9 glicocole 97.6 proline 96.6 serine 99.9 tyrosine 95.4 almandine 98.1 phenylalanine 97.1 valine 100.6 leucine 94.3 isoleucine 96.0 treonine 97.1 lysine 93.8 (96 ± 4)% Selectivity of the method: Interference study The first type of interfering compounds studied were those containing sulphur atoms, because they can produce interferences in the nitrogen determination as can be seen in the previous section for amino acids containing S. Two compounds, sodium sulphide (S2- ) and mercaptoethanol (R-SH), without nitrogen content, were studied. As it was expected, both compounds lowered the chemiluminiscence signal. Sulphur interference was twice the mercaptoacetic interference, the slopes of the calibration graphs obtained were 3058 and 1850 L/mg S. The response for cysteine, amino acid with sulphur content, and its derivative N- acetyl-cysteine were due to the contribution of the both atoms, N and S. The experimental signals were around 100 % of the theoretical signals obtained bearing in mind the slopes of the calibration graphs of ammonium and mercaptoethanol. The interference caused by the R-S-R group is small as can be derived from the recovery given in the previous section for metionine. The concentration of biogenic thiols in natural waters have been found to be about 0.25 ppb [15], and the higher limit admitted for drinking water for H2S is 50 ppb [7], at these levels the sulphur containing compounds did not interfere in the reaction, giving the same signal that the corresponding to the blank solution.
- 273. 249 The second type of possible interfering compounds assayed was some metallic ions. Cr(III) and Co(II), at concentration levels of 0.5 ppm, do not interfere. Cu(II), at concentration level of 50 ppb (maximum allowed limit for natural waters), decreased 10% the chemiluminiscence signal. Other cations, at the maximum allowed concentration (Fe(III), at concentration level of 0.5 ppm, Cd(II), at concentration level of 5 ppb, and Hg(II), at concentration level of 1 ppb) do not either interfere. Kjeldahl treatment Kjeldahl treatment was applied to nitrogen-containing compounds that did not give reaction with hypochlorite, in order to study its reactivity after sample digestion. Triethylamine, urea, histidine and arginine were measured at a final nitrogen concentration of 1.5 mg/L after Kjeldahl treatment. Recoveries obtained can be seen in Table 5. They are all near 100%, so applying Kjeldahl digestion, these nitrogen compounds will also be measured. TABLE 5. Individual Percentage of recovery and Mean percentage of recovery found for standards of Triethylamine, urea, histidine, arginine of final Nitrogen concentration of 1.5 mg/L, and for mixtures of Methylamine, Pentylamine and Dimethylamine (final Nitrogen concentration of 1.5 mg/L) and of Methylamine, Pentylamine, Dimethylamine and Trimethylamine (final Nitrogen concentration of 1.8 mg/L), after Kjeldahl treatment. ANALYTE % Recovery Mean Recovery (% REC ± s) TEA 94.1 urea 103.5 histidine 114.9 arginine 109.6 MA + PeA + DMA 96.3 MA + PeA + DMA + TMA 99.7 (103 ± 8)% Moreover, aliphatic amine mixtures containing or not trimethylamine have been digested and distilled by Kjeldahl method. The recovery percentages for two mixtures containing a total nitrogen content of 1.5 mg/L or 1.8 mg/L of Nitrogen is shown in Table 5, with both values close to 100%. In addition, a protein, as it is albumine, was also analyzed. The results, before and after the Kjeldahl digestion were different. In this case, almost half of the nitrogen is not detected without acidic digestion. Analysis of real water samples Several real water samples were analysed. To validate the accuracy of the method, all samples were fortified to apply the standard additions method, and the percentages of recovery of nitrogen added were near 100% (Table 6). This table show
- 274. 250 that there were not matrix effect in these samples, and that sample Nitrogen content can be deduced directly from standards calibration curve. TABLE 6. Concentration of Nitrogen found in spiked real water samples (mg/L)(S1- Mineral water, S2-Tap water, S3-Irrigation ditch water 2, S4-lake water, S5-sea water, S6-Waste (residual) water, S7-Irrigation ditch water 1 and S8-Fountain water), and percentage of recovery of the fortified concentration at different concentration levels. Sample Added Concentration (mg/L N) Found concentration (mg/L N) % Recovery (Rec ± s) % 0.75 0.69 ± 0.04 92 ± 6 1.5 1.52 ± 0.05 101 ± 4S1 3 2.91 ± 0.08 97 ± 3 0.75 0.64 ± 0.07 86 ± 10 1.5 1.46 ± 0.02 97.4 ± 1.3S2 3 2.75 ± 0.08 92 ± 3 0.75 0.71 ± 0.03 95 ± 4 1.5 1.58 ± 0.03 105.2 ± 1.7S3 3 3.004 ± 0.018 100.1 ± 0.6 0.75 0.724 ± 0.018 97 ± 2 1.5 1.69 ± 0.04 112 ± 3S4 3 2.62 ± 0.04 87.4 ± 1.3 0.75 0.76 ± 0.04 101 ± 6 1.5 1.58 ± 0.05 105 ± 3S5 3 2.85 ± 0.03 95.0 ± 1.1 0.75 0.692 ± 0.017 92 ± 2 1.5 1.43 ± 0.08 96 ± 6S6 3 2.696 ± 0.004 89.86 ± 0.12 0.75 0.79 ± 0.03 105 ± 4 1.5 1.581 ± 0.013 105.4 ± 0.9S7 3 3.034 ± 0.007 101.1 ± 0.2 1.5 1.49 ± 0.10 99 ± 7 S8 3 2.91 ± 0.10 97 ± 3 In three of the samples assayed, mineral water (S1), tap water (S2) and irrigation ditch water 2 (S3), the nitrogen contents were under detection limit of the method. Sea water (S5), presented increase of the blank signal and then, can not be analysed by the FI method directly. In other four samples, lake water (S4), waste water (S6), irrigation ditch water (S7) and fountain water (S8), detectable nitrogen was found when direct measurement was made. Table 7 shows the nitrogen contents found by the direct measurement of these water samples, calculated from standards calibration curve. Nitrogen content on real water samples was also deduced by this methodology after Kjeldahl treatment. As can be deduced from results (Table 7), two groups of samples have appeared: the first one showed similar nitrogen concentrations before and
- 275. 251 after the Kjeldahl digestion (this is the case of S6 and S7). S6 was also analysed by the Nessler and o-phthaldialdehyde/N-Acetyl-L-Cysteine (OPA/NAC) methods for ammonium [16], and after Kjeldahl treatment, the found concentrations were (180 ± 3, n=3) and (166 ± 3, n=3) mg/L N for Nessler and OPA/NAC procedures respectively. For this type of samples, we could replace the tedious Kjeldahl treatment by the application of the direct simple FI system presented in this work. The second group of samples, showed different nitrogen concentrations before and after the Kjeldahl digestion (this is the case of S4, S5 and S8). The proposed method for S4 provided 60 % of the N-Kjeldahl, S5 presented increase of the blank signal and then can not be analysed by the FI method directly as mentioned previously. The results for S8 can be explained by the presence of nitrogen containing compounds that does not react completely with the hypochlorite, but are transformed into ammonium after Kjeldahl treatment or, more probably, by the presence of complex organic matrix, decomposed by the acidic digestion (in a similar way that albumine results predict). S8 was also analysed by the Nessler and OPA-NAC method for ammonium [16], the results were (0.25±0.02, n=3) and (0.29±0.02, n=3) mg/L N, respectively before Kjeldahl treatment, and (1.66 ± 0.02, n=3) and (1.635 ± 0.18, n=3) mg/L N, respectively after Kjeldahl treatment. These results are near those obtained with the proposed chemiluminescence FI detection procedure. TABLE 7. Total Nitrogen content (Organic and ammonium)(mg/L N) found in different water samples studied, before and after Kjeldahl treatment. Samples analysed in triplicate. Nitrogen Content (mg/L N) Sample Before Kjeldahl Treatment After Kjeldahl Treatment S4 (0.92 ± 0.15) (1.59 ± 0.06) S5 N.D. (1.66 ± 0.10) S6 (140 ± 8) (149 ± 11) S7 (0.52 ± 0.04) (0.57 ± 0.03) S8 (0.35 ± 0.05) (1.99 ± 0.07) CONCLUSIONS The response of amino compounds (-CH2-NH-) including ammonium by applying the proposed methodology gave recoveries near 100% for primary and secondary aliphatic amines, polyamines, arylalkylamines and fourteen of the aminoacids assayed. Tertiary amines and aromatic or resonant nitrogens did not give the reaction, N- proteins is incompletely recovered. The reaction was interfered by sulphide groups, but not at concentrations below their legislation level. Metal ions studied have not resulted interferences below their legislation levels. The method is capable of estimating total dissolved N-amino in a sample very quickly. Matrix effects have not been observed in fortified samples, and accurate results have been obtained with recoveries near 100%. Results shown in this paper permit to establish that for some kind of samples, Kjeldahl treatment can be omitted and the simpler FI system presented in this work used. This option is very advantageous because
- 276. 252 the method does not require sample treatment, three replicates of the sample can be made in 1 min, toxicity of the reagents is low and the method can be used in in situ monitoring . The proposed methodology can be also used as an alternative for final ammonium detection after Kjeldahl digestion of the samples. The precision of the method for ammonium determination was very good, with RSD of 0.3-3.7 % for intraday repeatibility values, and 1.9% for interday reproducibility of the slope calibration line values. These values were similar to Nessler method precision (%RSD=0.4%), and better than OPA-NAC method precision (%RSD below 12.5%). Dynamic range was 0.24-4 mg/L N. For Nessler method, it was 0.66-3.88 mg/L N and for OPA-NAC method it was 0.155-1.088 mg/L N. Detection limit reached with the proposed method (0.07 mg/L N) was similar to that obtained with OPA-NAC method (0.054 mg/L N), and better than that reached with Nessler reference method (0.46 mg/L N). The rapidity of the proposed method resulted excellent (60 samples/hour) compared with Nessler (6 samples/hour) or OPA/NAC (10 samples/hour) methods. ACKNOWLEDGEMENTS The authors are grateful to the Ministerio de Ciencia y Tecnología of Spain for financial support received for the realization of Project BQU2003-06138 . Susana Meseguer Lloret expresses her gratitude to Ministerio de Educación y Cultura (Spain) for the predoctoral grant. BIBLIOGRAPHY [1] T.P. Burt, A.L. Heathwaite, S.T. Trudgill. Nitrate: Processes, Patterns and Management. John Wiley & Sons, New York 1993. [2] E.-S. A. Badr, E.P. Achterberg, A.L. Tappin, S.J. Hill, C.B. Braungardt, Trends Anal. Chem. 22 (2003) 819. [3] K.Cameron, C.Madramootoo, A.Crolla, C.Kinsley. Wat. Res. 37 (2003) 2803. [4] S.E.Tsiouris, A.P.Mamolos, K.L.Kalburtji, D.Alifrangis,Agric. Ecosyst. & Envir., 89 (2002) 117. [5] S.H. Ensign, M.A. Mallin, Wat. Res., 35 (2001) 3381. [6] V. P. Aneja, B. Bunton, J. T. Walker, B. P Malik, Atm. Environ., 35 (2001) 1949. [7] Directive 98/83/CEE. [8] B.M. Jones, C.G. Daughton, Anal. Chem. 57 (1985) 2320. [9] EPA-821-R-01-004. Method 1687. 2001 [10] G. Alavoine, B. Nicolardot, Analusis 28 (2000) 88. [11] J. H. Sharp et al., Marine Chemistry 78 (2002) 171. [12] J. Li, P. Purnendu, K. Dasgupta, Anal. Chim. Acta 398 (1999) 33. [13] P.Campins-Falcó, L.A.Tortajada-Genaro, F.Bosch-Reig, Talanta. 55 (2001) 403- 413. [14] Application Note ASN 52/85. Tecator Manual Digestion system 1007 (1985). [15] D. Tang, L.S. Wen, P.H. Santschi, Anal. Chim. Acta, 408 (1-2) (2000) 299-307. [16] S.Meseguer-Lloret, C.Molins-Legua, P.Campíns-Falcó, Intern. J. of Environ. Anal. Chem. 82 (7) (2002) 475-489.