





















































Analysis of elemental impurities in API
- 1. Implementation of Elemental Impurities in APIs Dr. A. Amsavel
- 2. Content • Introduction • Guidelines on elemental impurities • Background for new requirement • Standard / Limit • Procedure & instrument • Method development & validation • Implementation
- 3. General Notices USP 38—NF 33 • General chapters <232> was published June 1, 2012, in the Second Supplement to USP 35 to effect from Feb 1, 2013 and deferred. • As per USP37 Sup-2, General Notices provision becomes official from January 1, 2018. – Elemental impurities has to be controlled in official drug products according to the principles defined and requirements standard specified in Elemental Impurities—Limits <232>, Also see <232> for information related to drug substances and excipients. • But It is expected for drug substances and excipients.
- 4. USP on Elemental Impurities USP General Chapter <231> Background and issues Toxicology considerations Chapter <232> - Elements and Limits Chapter <233> - Procedures Implementation Plans
- 5. Guidelines 1. Guideline On The Specification Limits For Residues Of Metal Catalysts Or Metal Reagents ; EMEA/CHMP/SWP/4446/2000; 21 February 2008 2. ICH Guideline- Q3D on Elemental Impurities (Step-4; 16 December 2014) 3. CDER adopted ICH: Q3D Elemental Impurities-Guidance for Industry; September 2015 4. EMA adopted ICH :EMA/CHMP/ICH/353369/2013- 25 August 2015
- 6. Why this new requirement Presently, there is a test for Heavy metals; then why regulatory/ standard setting agencies are insist for revision Is this change required? • As a professional in Industry • As consumer
- 7. Disadvantages of Heavy metal test: Presently Heavy metal test is being followed, but there are disadvantages : • Difficulties in reproducibility – Colour of solution is subjective, – standards change with time; – recovery issues • Difficulties with reagents – safety issues –All procedures generate H2S (including Thioacetamide) and H2S more toxic than cyanide –Thioacetamide is not allowed in several countries • Non-discriminatory screening test –Not element specific –Sensitivity varies by element –Only a few elements respond at required sensitivities
- 8. Disadvantage of Heavy Metals test
- 9. Key Elements under heavy metals • As, Cd, Pb, Hg are clasifed as key /major elements under heavy metals • Heavy metals are classified based on toxicity • ICH classified as class-1, 2 & 3
- 10. Toxicology • Approach to elemental impurity control that is both health based and risk based • Control metals that are toxic • Setting limits that are toxicologically relevant • At all times during a drug product’s shelf life • Risk-based approach -what is to be tested and when to test
- 11. Dose Levels and Toxicity –NOEL: No-Observed Effect Level –NOAEL: No-Observed-Adverse Effect Level –LOAEL: Lowest-Observed-Adverse Effect Level –MTD : Maximum Tolerated Dose –LD50: Lethal Dose to 50% of population Increasingdose Safe Less safe
- 12. Reference Dose (RfD) (toxic level) • An estimate of the daily dose of a chemical that will avoid toxic effects other than cancer • NOAEL or LOAEL is adjusted by uncertainty factors (UF) to allow for differences in sensitivity to chemicals –Human data UF = 10 –Animal data UF: –100 (NOAEL) –1000 (LOAEL) –1000 (NOAEL, less data)
- 13. PDE Calculation RfD = NOAEL/UF Eg. NOAEL is 10 μg/kg/day based on human data UF=10: RfD = 10μg/kg/day (NOAEL)/10 (UF) = 1μg /kg/day RfD is used to calculate Permissible Daily Exposure (PDE) (RfD X Weight) = PDE PDE Example: 50 kg person and RfD = 1 μg/kg/day: PDE = 1 μg/kg/day X 50 kg = 50 μg/day
- 14. PDE Calculation What if only LOAEL or limited NOAEL is available RfD = NOAEL/UF Eg.: NOAEL = 10 μg/kg/day based on animal data UF=1000: RfD = 10 μg/kg/day (NOAEL)/1000(UF) = 0.01 μg/kg/day RfD is used to calculate Permissible Daily Exposure (PDE) (RfD X Weight) = PDE PDE Example: 50 kg person and RfD = 0.1 μg/kg/day: PDE = 0.1 μg/kg/day X 50 kg = 0.5 μg/day
- 15. Elemental Impurities-Limits <232> • Implementation of Chapters <232> and <223> • Omission of Chapter <231> Heavy Metals • Elemental Impurities: – Scope and application – Elemental Impurities • Implementation : – Drug Product Analysis Option – Summation Option – Individual Component Option
- 16. General Notices USP 37—NF 32 Suppl- 2 • “Elemental impurities to be controlled in official drug products according to the principles defined and requirements standard”. Scope: • This General Chapter specifies limits for the amounts of elemental impurities in drug products. • The limits presented in this chapter do not apply to excipients and drug substances, except where specified in this chapter or in the individual monographs. However, elemental impurity levels present in drug substances and excipients must be known and reported.
- 17. General Chapter <232> Basics • Applies to drug products – Drug substances – Excipients • Does not apply to dietary supplements • Does not apply to veterinary products • Does not apply to Dialysates and Total Parenteral Nutritions (TPN) • Does not apply to vaccines • Speciation is not addressed in this Chapter • Procedures are specified in <233>
- 18. Elemental Impurities compliance Options available for determination of drug product: • Drug Product Analysis Option • Summation Option • Individual Component Approach for LVP Drug Product Analysis Option is Generally Applicable / well acceptable Determination & Calculation: –The results obtained from the analysis of a typical dosage unit scaled to a maximum daily dose. –Daily Dose PDE > measured value (μg/g) x maximum daily dose (g/day)
- 19. Source of Elemental Impurities • Environmental contaminants • Catalysts They may be: – Naturally occurring – minerals , Hyflo – Added intentionally - Catalyst – Introduced inadvertently –Through interactions with processing equipment –Through non-GMP routes - Reagents , early stage material • Elemental Impurities are – known to be present, – have been added, or – have the potential for introduction, – A risk-based control strategy may assure compliance. – compliance with the limits is required for all drug products at all times.
- 20. Default Concentration Limits for Drug Substances Element Concentration Limits (mg/g) for Oral Drug Products with a Maximum Daily Dose of £10 g/day Concentration Limits (mg/g) for Parenteral Drug Products with a Maximum Daily Dose of £10 g/day Concentration Limits (mg/g) for Inhalational Maximum Daily Dose of £10 g/day Cadmium 2.5 0.25 0.15 Lead 0.5 0.5 0.5 Inorganic arsenic0.15 0.15 0.15 Inorganic mercury1.5 0.15 0.15 Iridium 10 1.0 0.15 Osmium 10 1.0 0.15 Palladium 10 1.0 0.15 Platinum 10 1.0 0.15 Rhodium 10 1.0 0.15 Ruthenium •10• (ERR 1-Oct-2012)•1.0• (ERR 1-Oct-2012)•0.15• (ERR 1-Oct-2012) Chromium —a —a 2.5 Molybdenum 10 1.0 •1.0• (ERR 1-Oct-2012) Nickel 50 5.0 0.15 Vanadium •10• (ERR 1-Oct-2012)•1.0• (ERR 1-Oct-2012)•3.0• (ERR 1-Oct-2012) Copper 100 10 •10• (ERR 1-Feb-2013)
- 21. Classification as per ICH Q3D • Class 1: The elements, As, Cd, Hg, & Pb, are human toxicants; testing should only be applied when the risk assessment identifies • Class 2: Elements in this class are generally considered as human toxicants are route-dependent; – Class 2A elements have relatively high probability of occurrence in the drug product. Elements are: Co, Ni and V. – Class 2B elements have a reduced probability of occurrence in the drug product related to their low abundance and low potential – they may be intentionally added during the manufacture. Elements : Ag, Au, Ir, Os, Pd, Pt, Rh, Ru, Se and Tl. • Class 3: The elements in this class have relatively low toxicities. The elements in this class include: Ba, Cr, Cu, Li, Mo, Sb, and Sn. • Asses if high PDEs, generally > 500 μg/day.
- 22. Elements to be Considered in the Risk Assessment-ICH Element Class If intentionally added (all routes) Oral Parenteral Inhalation Cd 1 yes yes yes yes Pb 1 yes yes yes yes As 1 yes yes yes yes Hg 1 yes yes yes yes Co 2A yes yes yes yes V 2A yes yes yes yes Ni 2A yes yes yes yes Tl 2B yes no no no Au 2B yes no no no Pd 2B yes no no no Ir 2B yes no no no Os 2B yes no no no Rh 2B yes no no no Ru 2B yes no no no Se 2B yes no no no Ag 2B yes no no no Pt 2B yes no no no Li 3 yes no yes yes Sb 3 yes no yes yes Ba 3 yes no no yes Mo 3 yes no no yes Cu 3 yes no yes yes Sn 3 yes no no yes Cr 3 yes no no yes If not intentionally added
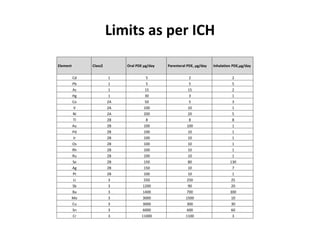
- 23. Limits as per ICH Element Class2 Oral PDE μg/day Parenteral PDE, μg/day Inhalation PDE,μg/day Cd 1 5 2 2 Pb 1 5 5 5 As 1 15 15 2 Hg 1 30 3 1 Co 2A 50 5 3 V 2A 100 10 1 Ni 2A 200 20 5 Tl 2B 8 8 8 Au 2B 100 100 1 Pd 2B 100 10 1 Ir 2B 100 10 1 Os 2B 100 10 1 Rh 2B 100 10 1 Ru 2B 100 10 1 Se 2B 150 80 130 Ag 2B 150 10 7 Pt 2B 100 10 1 Li 3 550 250 25 Sb 3 1200 90 20 Ba 3 1400 700 300 Mo 3 3000 1500 10 Cu 3 3000 300 30 Sn 3 6000 600 60 Cr 3 11000 1100 3
- 24. Calculation –The results obtained from the analysis shall be calculated to maximum daily dose. –Daily Dose PDE > measured value (μg/g) x max. daily dose (g/day) Example 1. 500 mg Tablet used Once a day dosing . Measured content of arsenic of 0.75 μg/g of tablet contents 1.5 μg/day (PDE limit) > 0.75 μg/g x 0.5 g/day x 1 (0.375 μg/day) 2. 1g Tablet used thrice a day dosing . Measured content of lead 2μg/g of tablet contents Is Tablet meet the requirement or fails
- 25. Compendial Procedures • Procedure 1: Can be used for elemental impurities generally amendable to detection by ICP-AES/OES • Procedure 2: Can be used for elemental impurities generally amendable to detection by ICP-MS. • Verification: Meets Procedure Validation Requirements
- 26. Implementation of Elemental Impurities Why <231> cannot be validated Choosing the appropriate instrument Qualifying the lab Determining which elements need to be validated Setting specifications Method development Method validation
- 27. Elemental Impurities - Procedures <233> Definitions Compendial Procedures –Procedure 1: ICP-OES –Procedure 2: ICP-MS Validation –Limit Procedures –Quantitative Procedures Calculations and Reporting
- 28. Analysis of Metal Residues as per EP 2.4.20 • Describes general approach for determination of metal catalyst or metal reagent residues in substances for pharmaceutical use. • System suitability needs to be demonstrated • Validation required for non-monograph procedures • Sample preparation, detection limit technique, instrument parameters is responsibility of the user • Allows AAS, XRF, ICP-AES, ICP-MS
- 29. Analysis of Metal Residues as per EP 2.4.20 • Sample Preparation – Aqueous of dilute nitric acid solutions – Hydrogen peroxide, hydrochloric acid, sulfuric acid, perchloric acid, hydrofluoric acid – Dilute bases – Organic solvents – Digestions • Hot-plate • Microwave-assisted digestions • Can be open-vessel if supported by spiking studies • Acceptance criterion for preparation of sample solution: a clear solution is obtained.
- 30. Analysis of Metal Residues as per EP 2.4.20 Measurement –Acceptance criteria for measurement system: measured concentration of standard solution does not differ from actual concentration by NMT 20% Quantitative Validation –Specificity –Range –Accuracy – Recovery –Repeatability –Intermediate Precision –Limit of Quantification –Limit of Detection (Limit test only)
- 31. Elemental Impurities – Procedure USP <233> Sample Preparation: • Neat or dilute as per analysis range • Direct aqueous solution – Sample is soluble in dilute acids and bases • Direct organic solution – Sample is soluble in an organic solvents • Indirect solution/Closed vessel digestion – Digestion is required using concentrated acids • Incomplete digestion: should dissolve majority of sample – Supported by spiking – if after filtration or centrifuge, recoveries are within limits set in USP <233> – Prepare appropriate ref (SRM) to matrix, values should be within COA • Leachate extraction can be justified • Total metal extraction is preferred
- 32. Instrument for Elemental analysis • ICP-AES • ICP-OES • ICP-MS
- 33. ICP-OES and ICP-AES Advantages –Dynamic range: 10^6-10^8 –Ability to analyze multiple elements at one time –Ability to perform a scan –Able to analyze samples with high dissolved solids without significant signal suppression –Precision: RSD < 5% Disadvantages –Higher detection limits –Spectral interferences –Consumes 2 – 5 mL of sample preparation
- 34. Elemental Impurities – Procedure Procedure I: ICP-AES, ICP-OES and Procedure II: ICP-MS –Standardization solution 1: 1.5J of the Target elements in a Matched matrix –Standardization solution 2: 0.5J of the Target elements in a Matched matrix –Prepare a matrix matched blank –Sample solution: Prepare/dilute the sample to obtain a final concentration of Target elements at 1.5J. J = The concentration (w/w) of the element t at the Target limit, appropriately diluted to the working range of the instrument. Note: Dilutions will be required if sample concentration exceeds 1.5J. Analyze in accordance with manufacturer’s suggestions –System suitability requirements – Tuning the instrument –Suggested wavelengths
- 35. Elemental Impurities – Procedure Procedure I: ICP-AES, ICP-OES and Procedure II: ICP-MS Analysis: • Standardization standard solution 1, Standardization standard solution 2, and Blank • Evaluate system suitability (after tuning) Note: Confirm the standards prepared correctly? & background of the instrument clean enough for use • If system suitability has been met – Analyze the Sample solution(s) • –Drift: Analyze standardization solution 1 (1.5J) after Sample solutions • Suitability criteria: NMT 20% drift for each Target element
- 36. Method Development Sample preparation used should yield a clear solution if at all possible • If solubility of sample is known, dissolve & dilute to get desired level • If solubility is unknown: • –Dissolve in dilute acid solution (1 – 5%) / Dissolve in organic solvent – Ensure solvent is not contaminated with elements of interest • Digest the sample in an open-vessel / Hot block or microwave – Needs to be supported by spiking studies to address possible loss of volatile elements • Digest samples in a closed-vessel microwave – Digestion tubes may need to be pre-screened prior to use – Keep acid concentration low in sample prep, if possible. • suggest <5% for nitric, hydrochloric and <2% for hydrofluoric • Dissolve in dilute base solution (<10%) – Stability of metals needs to be established – Bases may not be appropriate for all elements
- 37. Method Development Prepare sample at a dilution to the desired level – Example: Specification is 0.5 μg/g for Pb – Limit Test • Instrument detection limit is 0.1 μg/L • Detection limit of 0.25 μg/g is needed • A maximum dilution factor of 2500x will be required – Example: Specification is 0.5 μg/g for Pb – Quantitative Method Validation • Instrument detection limit is 0.1 μg/L • Detection limit of 0.125 μg/g is needed (based on 50% spike at 0.25 μg/g) • A maximum dilution factor of 1250x will be required
- 38. Method Development • Prepare a sample, replicates, spike, spiked replications; use individual sample portions for each preparation – Spike the matrix spike and matrix spiked replicates at the specifications prior to sample preparation – If spiking for a wide range of specifications (ex. 0.5 μg/g – 100 μg/g) • Prepare sample to obtain detection limits low enough for 0.5 μg/g • Dilute this sample preparation to bring the 100 μg/g spike within the linear range of the standards • Spike recoveries need to be: – Limit test: 85 – 115% and RSD < 20% between sample and spike preparations – Quantitative method validation: 70 – 150% and RSD < 20% between sample and spike preparations • LOQ satisfied by meeting the Accuracy requirements • Internal standard responses should not be suppressed or enhanced – Recoveries of 50 -125% are suggested, but may vary based on specific method – May need to evaluate sample preparation and use an alternative prep or technique
- 39. Method Development Decide on a limit test or a quantitative method validation, if not specified in monograph Limit test – Applicable when the elements of interest are not detected or trend at levels well below the specification – Raw materials where catalysts are not used in manufacturing Quantitative method validation – Applicable when elements of interest trend above the limit of quantitation, especially when they are close to the specification – Data needs to be trended • Draft a method validation protocol for review by all parties involved
- 40. Method Validation • Limit Procedures – Detectability – Precision (Repeatability) – Specificity • Quantitative Procedures – Accuracy – Precision – Repeatability – Ruggedness (Intermediate Precision) – Specificity – Limit of Quantitation, Range, and Linearity—satisfied by meeting Accuracy requirement
- 41. Method Validation Limit Procedures Detectability • Spiked sample solution 1: Prepare sample: – Spike at the target concentration (100%) for each target element – Spike prior to sample preparation – Analyze in triplicate • Spiked sample solution 2: Prepare sample: – Spike at the 80% of target concentration for each target element – Spike prior to sample preparation • Standard solution: Prepare working standard for the target element(s) at target concentrations – Analyze in triplicate • Unspiked sample solution: Prepare an unspiked sample aliquot Acceptance criteria: average of triplicate measurements of Spiked sample solution 1 is within ±15% of the average of triplicate measurements of Standard solution
- 42. Method Validation Limit Procedures • Precision (Repeatability) Sample solution (spiked) Prepare: – Six independent sample preparations – Spike each at the target concentration (100%) for each target element – Spike prior to sample preparation Acceptance criteria: RSD NMT 20% for each Target element • Specificity: – Unequivocally assess each Target element in the presence of components that may be expected to be present, including other Target elements, and matrix components
- 43. Method Validation • Limit Procedures • Specificity (example ICP-OES, ICP-AES) – Spectral interferences may be caused by background emission from continuous or recombination phenomena, stray light or overlap of spectral lines from another element or unresolved overlap of molecular band spectra. "subarray" on the CID detector. – A metals screen/scan analysis performed on this material demonstrated that the sample does/does not contain appreciable concentrations of any element that are likely to cause interferences on the analytes of interest. Discuss specific elements with the potential to cause interference and how they will be monitored or addressed.
- 44. Method Validation Quantitative Procedures • Accuracy – Standard solution: Prepare working standard for the elements of interest at target concentrations ranging from 50% to 200% of J – Test solution: Prepare sample: • Use the method developed • Spike at target concentrations of 50% to 200% of J for each target element • Prepare three replicates at each concentration • Spike prior to sample preparation – Acceptance criteria: 70% - 150% for the mean of three replicate preparations at each concentration
- 45. Method Validation Quantitative Procedures Precision (Repeatability) • Test solution: Prepare sample: – Six independent sample preparations – Spike at target concentrations (J) for each target element – Spike prior to sample preparation • Acceptance criteria: RSD NMT 20% for each Target element Precision (Ruggedness) • Perform Repeatability analysis over three independent events using the following events or combinations thereof: – on different days, or – with different instrumentation, or – with different analysts • Acceptance criteria: RSD NMT 25% (N=12) for each Target element
- 46. Method Validation Quantitative Procedures • Specificity (refer to slide for limit test) • Limit of Quantitation, Range, and Linearity –Demonstrated by meeting the Accuracy requirements (at 0.5 J)
- 47. Establishing In-house Lab Purchase appropriate instrumentation Purchase apparatus to assist in sample digestion • Hot block with polypropylene disposable digestion vessels : 50 mL / 15 mL – limited sample size available • Microwave system with digestion vessels : 50 mL /15 mL – Quartz – most appropriate for screen – Teflon – resistant to hydrofluoric acid – Borosilicate – disposable, may not be clean enough for trace levels Purchase – High Purity Acids (nitric, hydrochloric, hydrofluoric, sulfuric, perchloric) – Hydrofluoric, Perchloric –Bases – Ammonium hydroxide , Sodium hydroxide ., CFA-C, TMAH
- 48. Preparedness for implementation • Standards (NIST traceable) – Single element for spiking at varying specifications – Multi-element – Custom multi-element for specification levels – Single element standards for internal standards – Second source standards for verification • Plastic bottles or vials for sample preparation • Gases : Argon, hydrogen, helium gases • Highest purity – Ultrapure grade , Low krypton content – Use stainless steel gas lines, avoid aluminum and copper
- 49. Preparedness for Contract Lab Audit the lab for cGMP compliance/FDA / appropriate approvals; • Each material shall require its own validation • System suitability can be established with daily tune for ICP-OES, • ICP-OES and ICP-MS have a wide dynamic range, single standard analysis at 0.5J and 1.5J are sufficient, do not need to analyze 5 standards • Not all elements listed in the chapters will need to be validated for every material – As, Cd, Pb, Hg needs to be validated, at a minimum – If catalysts are not used validation is not required – Strongly suggest incorporating ICH Class 2A elements in minimum evaluation, as well
- 50. Preparedness for Contract Lab • Audit the contract lab for suitability of resources and compliance • Provide all information related to product • Solubility , handling & storage • Set specifications, work appropriate dilution/ concentration • Ensure to full metals scan at time of method development, so that • Communicate which elements require method validation based on risk assessment • Submit samples in plastic bag/containers to avoid metal contaminant ion • Ensure sufficient samples for development & method validation • Prepare or review and approve method validation protocol and Review and approve the report.
- 51. Summary Heavy Metals <231> must be replaced USP chapters <232> and <233>, Elemental Impurities – Limits <232> –Toxicological basis for limits –Options to determine compliance –Limits (aligned to ICH- Q3D) Elemental Impurities – Procedures <233> –Two compendial procedure –Limit Test and Quantitative procedures – Sample preparation – Method development – Method validation Working with contract Lab
- 52. Q&A 1. What are the elements need to be shown in the analysis 1. Big Four 2. Catalysts used in the process 3. All the elements present in <232> 4. Elements present as per the process only 5. Elements present in the drug product as per the process
- 53. Q&A 2) In the manufacturing process, there are no catalysts used and there is no possibility of any elemental impurity present. What are the elements to be shown in the analysis? 1. Big Four 2. All the elements present in <232> 3. Elements present in the drug product as per the process 4. No element is required
- 54. Q&A 3) What is the preferred way to determine the elemental impurities in the drug product. 1. Analysis of drug product 2. Individual components of product 3. Both 4. None
- 55. Answer • Q-1 – Ans: (1), (2), (5) • Q-2 – Ans: (1), (3) • Q-3 – Ans: (1)